



Maeshima Group / Genome Dynamics Laboratory
Liquid-like chromatin in the cell: What can we learn from imaging and computational modeling?
Yuji Itoh, Esmae J. Woods, Katsuhiko Minami, Kazuhiro Maeshima, and Rosana Collepardo-Guevara
Current Opinion in Structural Biology 71, 123-135 (2021) DOI:10.1016/j.sbi.2021.06.004
The loopy world of cohesin.
Kazuhiro Maeshima and Shiori Iida
eLife 10, e71585 (2021) DOI:10.7554/eLife.71585
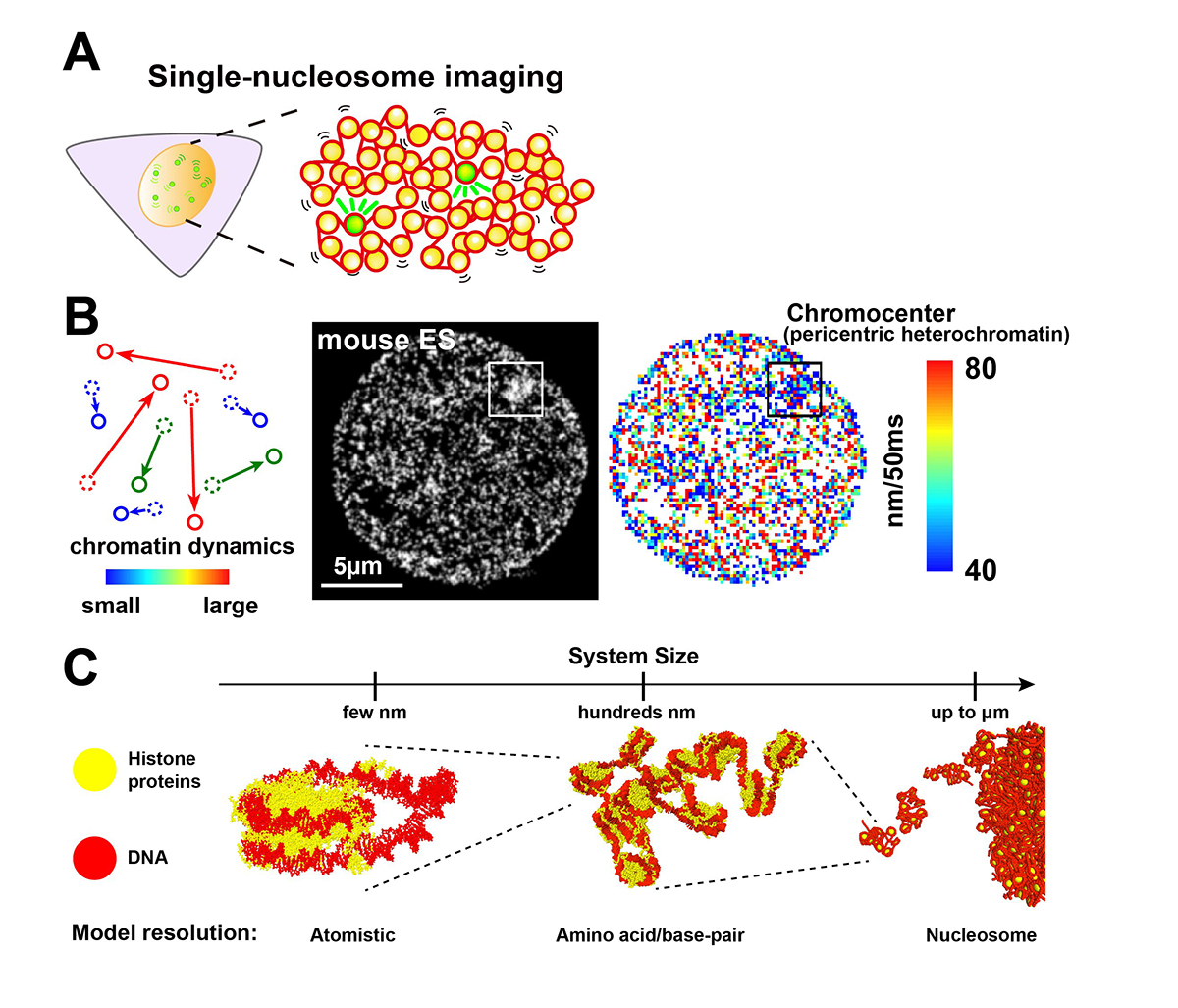
Chromatin in eukaryotic cells is a negatively charged long polymer consisting of DNA, histones, and various associated proteins. With its highly charged and heterogeneous nature, chromatin structure varies greatly depending on various factors (e.g., chemical modifications and protein enrichment) and the surrounding environment (e.g., cations): From a 10-nm fiber, a folded 30-nm fiber, to chromatin condensates/droplets. Recent advanced imaging such as single-nucleosome imaging (Figure 1A) has observed that chromatin exhibits a dynamic liquid-like behavior and undergoes structural variations within the cell (Figure 1B). Current computational modeling has made it possible to reconstruct the liquid-like chromatin in the cell by dealing with a number of nucleosomes on multi-scale levels, and has become a powerful technique to inspect the molecular mechanisms giving rise to the observed behavior, which imaging methods cannot do on their own (Figure 1C). Based on new findings from both imaging and modeling studies, we discuss the dynamic aspect of chromatin in living cells and its functional relevance.
This work was supported by JSPS grant (19K23735, 20J00572, 20H05936, 21H02453), the Takeda Science Foundation, the Uehara Memorial Foundation, NIG Postdoctoral Fellowship, JSPS Postdoctoral Fellowship (PD).
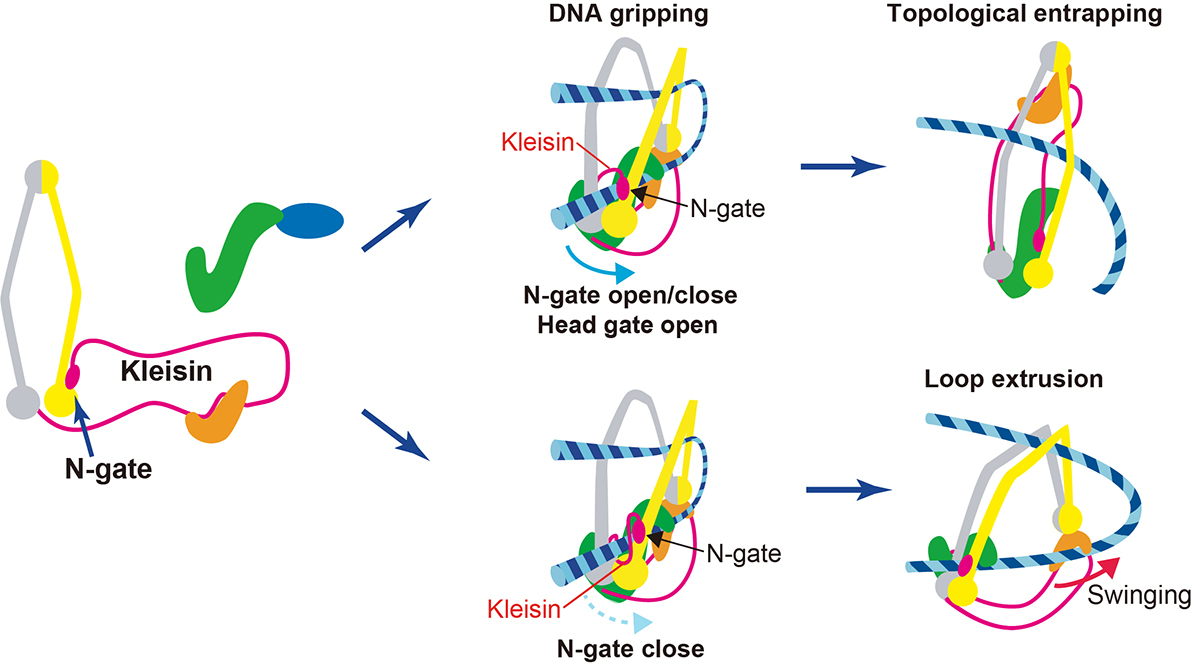
Also, on July 26th, a professor at Genome Dynamics Laboratory Kazuhiro Maeshima and SOKENDAI Ph.D. student Shiori Iida published an Insight paper in eLife. Chromatin higher-order structures, such as chromatin loop domains, are critical for chromatin to perform various functions in the cell. The formation of these chromatin loops is thought to be mediated by a ring-shaped molecular complex, cohesin (Figure 2, left). Currently, the mechanism of chromatin loop formation is a hot topic in cell biology, and a model called loop extrusion, in which cohesin pushes DNA out of the ring, has been attracting much attention. In fact, cohesin has been shown to extrude naked DNA in vitro. Recently, Dr. Frank Uhlmann and his colleagues have shown how cohesin excludes DNA loops in vitro (Figure 2, bottom)(Higashi et al., “A Brownian ratchet model for DNA loop extrusion by the cohesin complex”. eLife, 2021 DOI:10.7554/eLife.67530). Based on the Higashi et al. paper, Maeshima and Iida discussed the intracellular behavior of cohesin and whether the loop extrusion reported really occurs in the cell.
Press release

Estrogens influence female itch sensitivity via the spinal gastrin-releasing peptide receptor neurons
K. Takanami*, D. Uta, K. Matsuda, M. Kawata, E. Carstens, T. Sakamoto, and H. Sakamoto
*Corresponding author
PNAS 118, e2103536118 (2021) DOI:10.1073/pnas.2103536118
![]() Press release (In Japanese only)
Press release (In Japanese only)
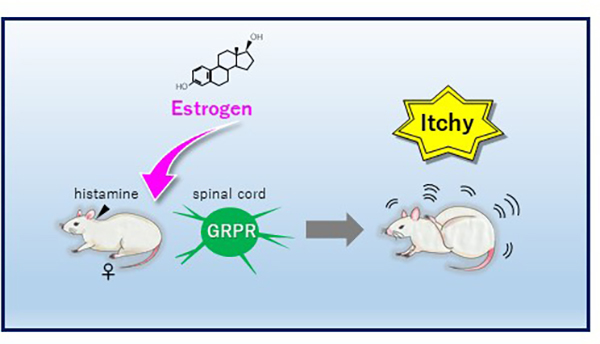
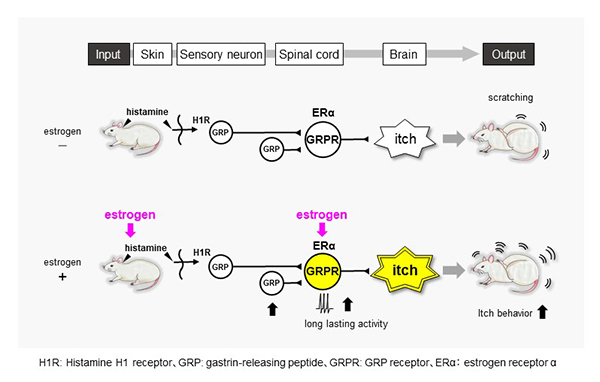
“Itch” sensitivity in women changes during periods when female sex hormones fluctuate, such as during pregnancy and menopause. Especially, many women exhibit itch symptoms during pregnancy, but the underlying mechanism of the change in itch sensitivity is unknown. Here Dr. Takanami et al. demonstrate that estradiol, but not progesterone, enhances histamine-evoked itch-related scratching behavior in female rats. This is associated with the enhancement of activity of “gastrin-releasing peptide receptor (GRPR)” neurons in the spinal cord (Fig. 1). The findings suggest that female hormone, estrogens selectively enhance histamine-evoked itching by regulating spinal GRP system in females (Fig. 2). This may account for why itch sensation varies across the female lifecycle and provides a novel basis for treating itchy diseases in females.
▶ This article was seleceted “In This Issue” of PNAS.

PZLAST: an ultra-fast amino acid sequence similarity search server against public metagenomes
H. Mori, H. Ishikawa, K. Higashi, Y. Kato, T. Ebisuzaki, K. Kurokawa
Bioinformatics 2021 July 7 DOI:10.1093/bioinformatics/btab492
![]() Press release (In Japanese only)
Press release (In Japanese only)
Similarity searches of amino acid sequences against the public metagenomic data can provide users insights about the function of sequences based on the environmental distribution of similar sequences. However, a considerable reduction in the amount of data or the accuracy of the result was necessary to conduct sequence similarity searches against public metagenomic data, because of the vast data size more than Terabytes. Here, we present an ultra-fast service for the highly accurate amino acid sequence similarity search, called PZLAST, which can search the user’s amino acid sequences to several Terabytes of public metagenomic sequences in approximately 10-20 minutes. PZLAST accomplishes its search speed by using PEZY-SC2, which is a MIMD many-core processor. Results of PZLAST are summarized by the ontology-based environmental distribution of similar sequences. PZLAST can be used to predict the function of sequences and mine for homologs of functionally important gene sequences.
Source: H. Mori, et al., Bioinformatics DOI: 10.1093/bioinformatics/btab492
▶ PZLAST is available here.
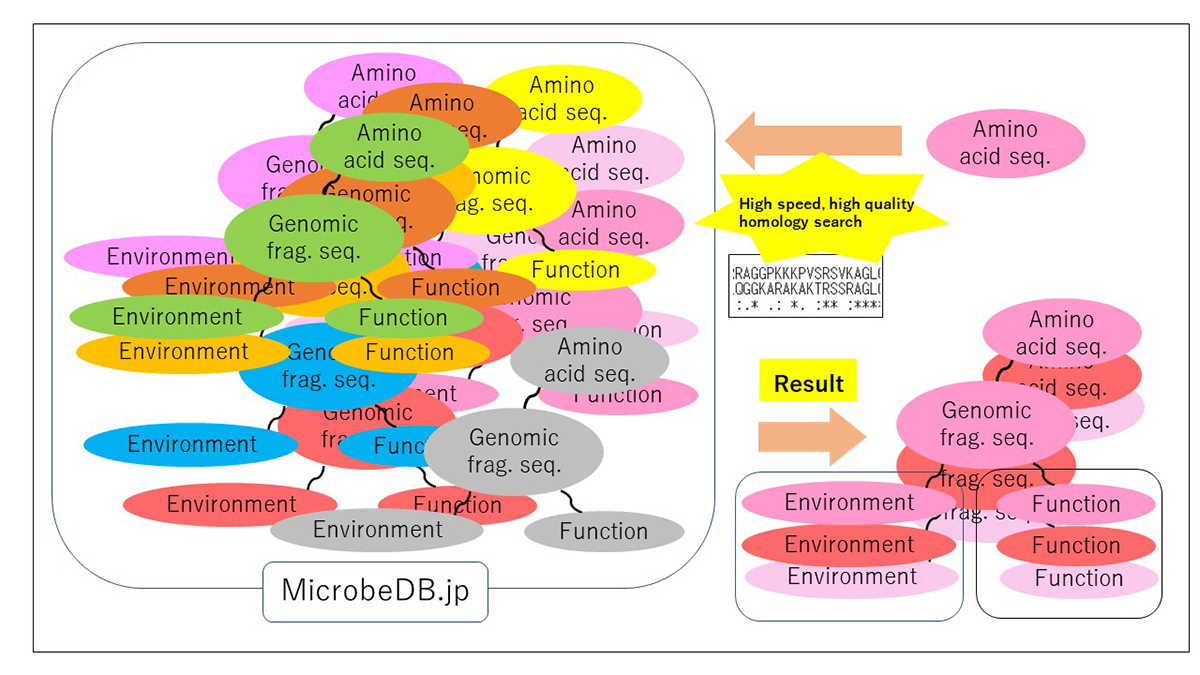
Press release

Internal microbial zonation during the massive growth of marimo, a lake ball of Aegagropila linnaei in Lake Akan
R. Nakai, I. Wakana, H. Niki
iScience 2021 June 12 DOI:10.1016/j.isci.2021.102720
Press release (In Japanese only)
Marimo (lake ball) is an uncommon ball-like aggregation of the green alga, Aegagropila linnaei. Although A. linnaei is distributed in fresh and brackish waters in the northern hemisphere, marimo colonies are found only in particular habitats. Here, we report the bacterial communities inside various sizes and aggregating structures of natural marimo collected from Lake Akan, Japan. We observed multi-layers composed of sediment particles only in the sizeable radial-type marimo with a >20 cm diameter, not in the tangled-type marimo. The deeper layers were enriched by Nitrospira, potential sulphur-oxidizing bacteria, and sulphate-reducing bacteria. Microorganisms of the multi-layers would form biofilms incorporating nearby sediment, which would function as microbial “seals” within large radial-type marimo. We propose that the layer structure provides habitats for diverse bacterial communities, promotes airtightness of the marimo, and finally contributes to the massive growth of the aggregation. These findings provide clues to deciphering the growth of endangered marimo.
Source: R. Nakai, et al., iScience DOI:10.1016/j.isci.2021.102720
Due to a network system error, the intermittent disconnection to our website happens.
We apologize for any inconvenience and appreciate your understanding and cooperation.
ネットワークシステムの不具合によりウェブサイトに断続的にアクセスしにくい事象が発生しています。
ご利用の皆様にはご迷惑をおかけして申し訳ございませんが、ご理解とご協力をお願い申し上げます。
There was a problem that NIG website was unavailable on Jun 12th. Currently, the problem has been restored.
We apologize for any inconvenience.
6月12日深夜から研究所ウェブサイトが閲覧できない不具合が発生していました。
6月15日午後4時現在、不具合は解消しています。
ご利用の皆様にはご迷惑をおかけしましたことをお詫び申し上げます。
There was a problem that NIG website was unavailable on Jun 11th. Currently, the problem has been restored.
We apologize for any inconvenience.
6月11日午後から研究所ウェブサイトが閲覧できない不具合が発生していました。
6月11日午後4時現在、不具合は解消しています。
ご利用の皆様にはご迷惑をおかけしましたことをお詫び申し上げます。
3D genomics across the tree of life reveals condensin II as a determinant of architecture type
Hoencamp, C., Dudchenko, O., Elbatsh, A. M.O., Brahmachari, S., Raaijmakers, J. A., van Schaik, T., Cacciatore, Á. S., Contessoto, V., van Heesbeen, R. G.H.P., van den Broek, B., Mhaskar, A. N., Teunissen, H., St Hilaire, B. G., Weisz, D., Omer, A. D., Pham, M., Colaric, Z., Yang, Z., Rao, S. S.P., Mitra, N., Lui, C., Yao, W., Khan, R., Moroz, L. L., Kohn, A., St. Leger, J., Mena, A., Holcroft, K., Gambetta, M. C., Lim, F., Farley, E., Stein, N., Haddad, A., Chauss, D., Mutlu, A. S., Wang, M. C., Young, N. D., Hildebrandt, E., Cheng, H. H., Knight, C. J., Burnham, T. L.U., Hovel, K. A., Beel, A. J., Mattei, P.-J., Kornberg, R. D., Warren, W. C., Cary, G., Gómez-Skarmeta, J. L., Hinman, V., Lindblad-Toh, K., di Palma, F., Maeshima, K., Multani, A. S., Pathak, S., Nel-Themaat, L., Behringer, R. R., Kaur, P., Medema, R. H., van Steensel, B., de Wit, E., Onuchic, J. N., Di Pierro, M., Lieberman-Aiden, E., Rowland, B. D.
Science 372, 984-989 (2021) DOI:10.1126/science.abe2218
An international collaborative team, which was led by B.D. Rowland at the Netherlands Cancer Institute and E. Lieberman Aiden (NIG International Strategic Advisor) at Baylor College of Medicine, investigated genome folding across the eukaryotic tree of life (24 species, Fig. B). The team found four types of 3D genome architecture at chromosome-scale (Fig. A). Each type appeared and disappeared repeatedly during eukaryotic evolution. The type of genome architecture that an organism exhibits correlates with the absence of condensin II subunits (Fig. B). Condensin is a protein complex required for mitotic chromosome assembly. In vertebrates, it is known that there are two types of condensins, condensin I and II, and that condensin II shortens mitotic chromosomes.
Moreover, the team demonstrated that condensin II depletion converted the architecture of the human genome to a state resembling that seen in organisms such as fungi or mosquitoes (Rabl-like, Fig. C). In this state, centromeres clustered together at nucleoli, and heterochromatin domains merged. The team proposed a physical model in which lengthwise compaction of chromosomes by condensin II during mitosis determines chromosome-scale genome architecture, with effects that are retained during the subsequent interphase. This mechanism is likely conserved since the last common ancestor of all eukaryotes.
The part to which NIG contributed was supported by MEXT KAKENHI grants (20H05936).
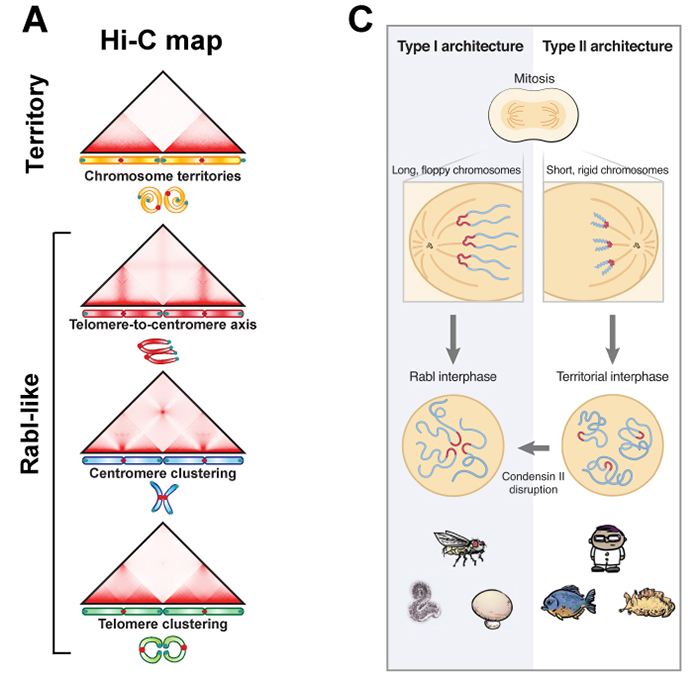
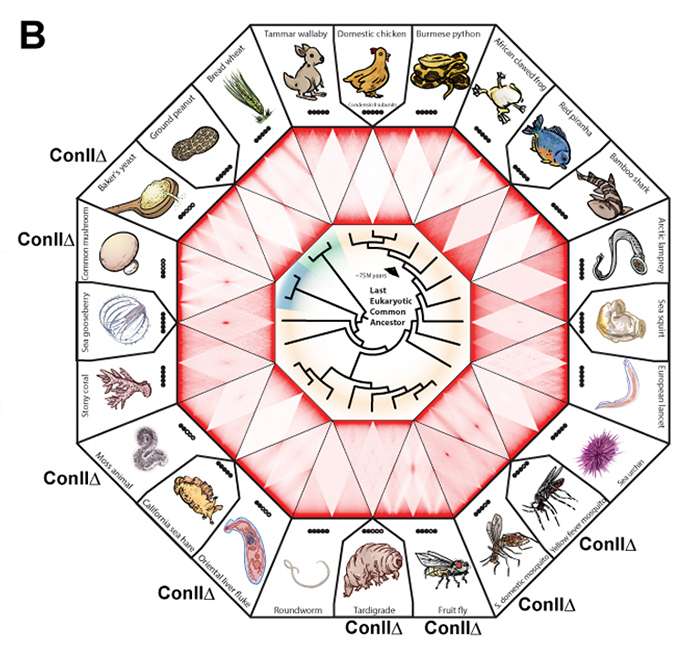
Figure: (A) Chromosome positioning in the nucleus classified by Hi-C maps: “chromosome territory” and “rabl-like”. (B) Hi-C maps of 24 species covering animals (yellow), fungi (blue) and plants (green). Presence of the condensin II subunits in each species is indicated by solid black circles. In case of condensin II-deficiency, shown as “Con IIΔ”. (C) Model. Having shorter chromosomes during cell division leads to separate centromeres and territorial genome architecture in the subsequent interphase. Reducing chromosome lengthwise compaction by condensin II disruption, leads to enhanced centromere clustering, loss of chromosome territories, and a Rabl-like genome architecture.
Our website will be under maintenance from 3:00 p.m. on May 27 to 9:00 a.m. on May 28. The website will be basically available during the maintenance period, but may be intermittently unavailable.
Thank you for your understanding and cooperation.
本日5月27日15:00より翌28日9:00まで、遺伝研ウェブサイトのメンテナンスを予定しています。メンテナンス時間においてもウェブサイトは利用できますが、断続的に利用できない場合があります。
皆様のご理解とご協力をお願いいたします。
Press release
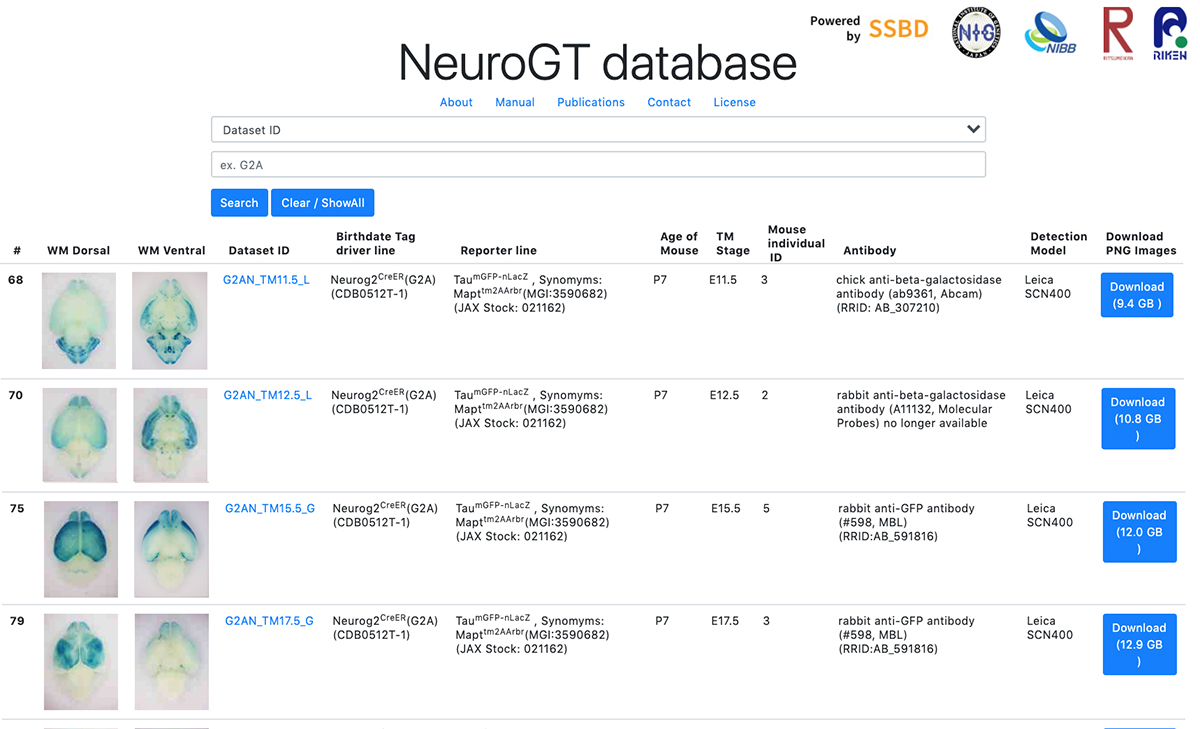
NeuroGT: A brain atlas of neurogenic tagging CreER drivers for birthdate-based classification and manipulation of mouse neurons
T Hirata*, Y Tohsato, H Itoga, G Shioi, H Kiyonari, S Oka, T Fujimori, S Onami *Corresponding author
Cell Reports Methods 1, 100012 (2021) DOI:10.1016/j.crmeth.2021.100012
Press release (In Japanese only)
Neuronal birthdate is one of the major determinants of neuronal phenotypes. However, most birthdating methods are retrospective in nature, allowing very little experimental access to the classified neuronal subsets. Hirata and her collaborators have developed four neurogenic tagging mouse lines that can assign CreER-loxP recombination to neuron subsets that share the same differentiation timing in living animals. Because this genetic tag is irreversible, the classified neuronal subset can be subsequently subjected to various experimental manipulations.
To encourage the use of this resource, Hirata et al. have launched “NeuroGT database”, a brain atlas of neurogenic tagging mouse lines, which includes holistic image data of the loxP-recombined neurons and their processes across the entire brain that were tagged at each single day during the neurodevelopmental period. Users can search for the datasets from a web browser using the terns in the meta-information, interactively view thumbnail images of the sections, and download the high-resolution section images.
The NeuroGT is now open to public, offering people the opportunity to find specific neurogenic tagging driver lines and the stages of tagging appropriate for their own research purposes. The driver mouse lines can be obtained from Riken Bioresource Center.
The NeuroGT database was constructed by joint efforts of different research groups; mouse engineering (RIKEN Center for Biosystems Dynamics Research), neuroscience (National Institute of Genetics), bioimaging (National Institute for Basic Biology), and image informatics (Ritsumeikan University, RIKEN Center for Biosystems Dynamics Research)
This research was supported by ROIS Challenging Exploratory Research Projects for the Future Grant and Grant-in-Aid for Publication of Scientific Research Results (Database, 19HP7002). High resolution digitization of section images was supported by Advanced Bioimaging Support (JP16H06280) and Grants-in-Aid for Scientific Research (20H03345, JP18H05412).
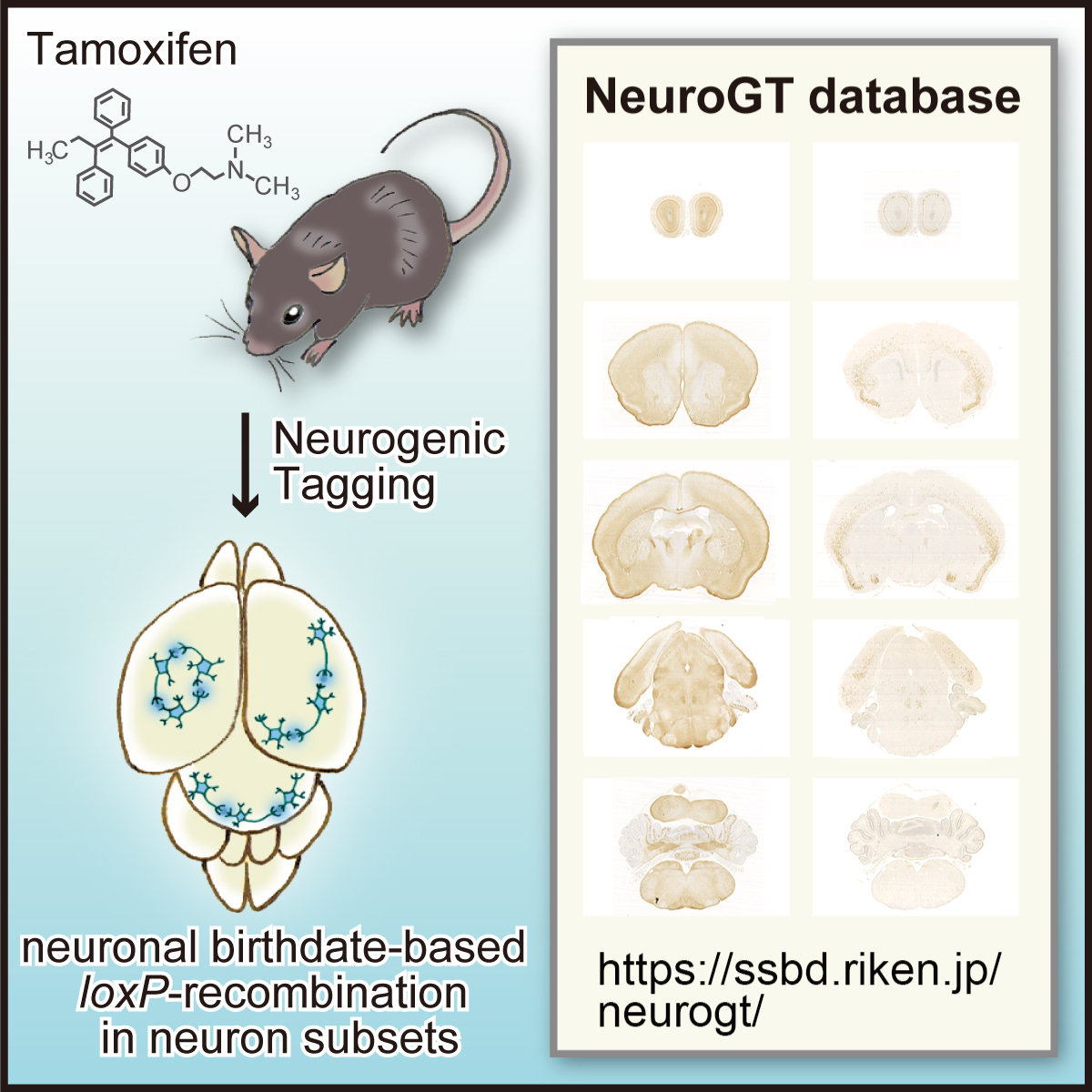
Figure: Graphical abstract of “NeuroGT Database”
Video: Thumbnail images of coronal sections can be sequentially viewed along the antero-posterior brain axis by dragging the slider. The images stained for the two different reporters, membrane-localized GFP and nucleus-localized-βGAL are stacked separately. The sync button automatically matches the section level of the two reporter images.
National Institute of Genetics has offered cooperation to Shizuoka prefecture and has signed a contract concerning investigation cooperation since April 2020. In response to the spread of infection of the new coronavirus variant in these days, a memorandum of cooperation on molecular epidemiological investigation (SARS-CoV-2 RNA whole genome analysis) of novel coronavirus has been signed between our institute and Heita Kawakatsu, Governor of Shizuoka Prefecture, at the government office.
The principles set in the memorandum are: “to support for proactive epidemiological investigation (SARS-CoV-2 RNA whole genome analysis) promoted by Shizuoka prefecture”, “to contribute to treatment of patients infected with novel coronavirus and prevention of the spread of the virus”, “to disclose molecular epidemiological information obtained from specimen samples in order to contribute to the conquest of COVID-19” and “to give due consideration to the protection of personal information when molecular epidemiological information are disclosed”. Based on this memorandum, our institute implements support for SARS-CoV-2 RNA whole genome analysis in earnest.
April 30th, 2021
National Institute of Genetics

Left, Heita Kawakatsu (Governor, Shizuoka Prefecture): Right, Fumio Hanaoka (Director-General, National Institute of Genetics)
Decoding the transcriptome of pre-granulosa cells during the formation of primordial follicles in the mouse
Kurumi Fukuda, Masafumi Muraoka, Yuzuru Kato, and Yumiko Saga
Biology of Reproduction 2021 April 13 DOI:10.1093/biolre/ioab065
6. Primordial follicles, a finite reservoir of eggs in mammalian ovaries, are composed of a single oocyte and its supporting somatic cells, termed granulosa cells. Although their formation may require reciprocal interplay between oocytes and pre-granulosa cells, precursors of granulosa cells, little is known about the underlying mechanisms. We addressed this issue by decoding the transcriptome of pre-granulosa cells during the formation of primordial follicles. We found that marked gene expression changes, including extracellular matrix, cell adhesion, and several signaling pathways, occur along with primordial follicle formation. Importantly, differentiation of pre-granulosa cells to granulosa cells was delayed in mutant ovaries of the germ cell-specific genes Nanos3 and Figla, accompanied by perturbed gene expression in mutant pre-granulosa cells. These results suggest that proper development of oocytes is required for the differentiation of pre-granulosa cells.
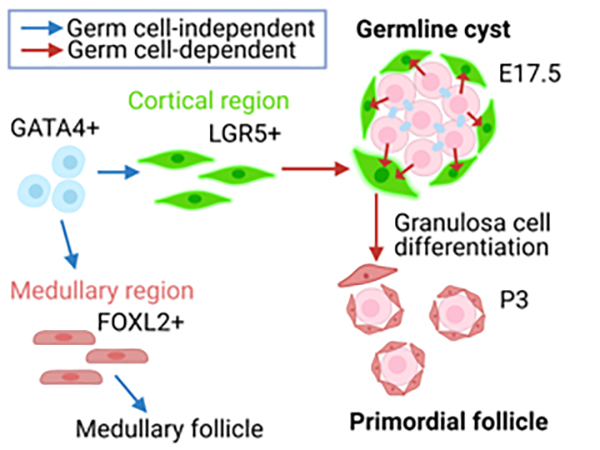
Figure: Pre-granulosa cells (Lgr5+) originate from GATA4 + precursors (blue arrow) in an oocyte-independent manner. On the other hand, differentiation of pre-granulosa cells to granulosa cells requires oocytes (red arrow).
Infection of novel coronavirus (SARS-CoV-2) variants is spreading mainly in large cities. In response to the spread of novel coronavirus (COVID-19), we, National Institute of Genetics, have offered cooperation to Shizuoka prefecture in April 2020 and have concluded an agreement on investigation cooperation in July.
We will start analyzing genome information (whole genetic information) of the specimens collected from COVID-19 patients in the prefecture as early as this month upon further cooperation request from Shizuoka prefecture. Specimen samples (nucleic acid substance) provided by the prefecture are not infectious and safe. We quickly determine presence/absence of variant virus by analyzing the genome information of the samples and report the result to the prefecture.
Genome sequencing of those samples enables us to accurately detect and identify SARS-CoV-2 variants spreading within the prefecture and to contributes to the prevention measures against COVID-19 by using genome sequence information.
SARS-CoV-2 genome sequence decoded by this research investigation will be released through the INSD (the International Nucleotide Sequence Database) and GISAID (Global Initiative on Sharing Avian Influenza Data). In the flamework of open science, our institute will actively cooperate with other projects on anti-virus measures conducted by the government.
April 26th, 2021
HANAOKA, Fumio
Director-General, National Institute of Genetics
Replication timing maintains the global epigenetic state in human cells
Kyle N Klein, Peiyao A Zhao, Xiaowen Lyu, Takayo Sasaki, Daniel A Bartlett, Amar M Singh, Ipek Tasan, Meng Zhang, Lotte P Watts, Shin-Ichiro Hiraga, Toyoaki Natsume, Xuemeng Zhou, Timour Baslan, Danny Leung, Masato T Kanemaki, Anne D Donaldson, Huimin Zhao, Stephen Dalton, Victor G Corces, David M Gilbert
Science 372, 371-378 (2021) DOI:10.1126/science.aba5545
The temporal order of DNA replication is conserved from yeast to humans, but its biological significance remains unclear. We eliminated the protein RIF1, a master regulator of replication timing, in several human cell lines. RIF1 loss during the G1 phase of the cell cycle resulted in a heterogeneous, nearly random replication timing program from the first S phase. Altered replication timing was followed by replication-dependent redistribution of active and repressive histone modifications and alterations in genome architecture. These results support a model in which replication timing orchestrates the epigenetic state of newly replicated chromatin.
The Kanemaki laboratory contributed to the generation of a conditional RIF1 mutant using the auxin-inducible degron system.
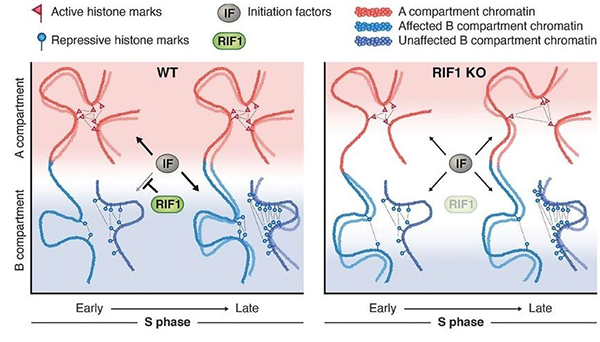
Figure: A model showing the role of the replication-timing program for the maintenance of the epigenome.
 Prof. Maeshima’s comment on the role of phase separation in transcription, “New door to understanding transcription, open with caution.” was published in the April 15 issue of Molecular Cell (Cell Press).
Prof. Maeshima’s comment on the role of phase separation in transcription, “New door to understanding transcription, open with caution.” was published in the April 15 issue of Molecular Cell (Cell Press).
![]() https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)00267-7
https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)00267-7
In recent years, a principle called “liquid-liquid phase separation (LLPS)” has been in the limelight in cell biology. It is thought that LLPS can increase the concentration of specific molecules in cells and create membrane-less structures “droplets”, making it possible to control cellular functions spatially and temporally. Because of this trend, many researchers claim that the intracellular assembly of various molecules is a droplet.
This time, while Prof. Maeshima admits that LLPS is an exciting new concept in cell biology, he argues that there is a lack of tools to prove the existence of “droplets” in the cell and that careful research based on the observation of individual molecule behavior is required.
Kakutani Group / Epigenomics Laboratory
The chromatin remodeler DDM1 prevents transposon mobility through deposition of histone variant H2A.W
Akihisa Osakabe, Bhagyshree Jamge, Elin Axelsson, Sean A. Montgomery, Svetlana Akimcheva, Annika Luisa Kuehn, Rahul Pisupati, Zdravko J. Lorković, Ramesh Yelagandula, Tetsuji Kakutani, and Frédéric Berger
Nature Cell Biology 23, 391-400 (2021) DOI:10.1038/s41556-021-00658-1
Transposable elements (TEs) are generally silenced by epigenetic modifications such as methylation of DNA and histone. An Arabidopsis chromatin remodeler protein DDM1 (Decrease in DNA methylation 1) was identified by genetic screening as a factor necessary for maintenance of these silencing marks. However, the underlying mechanisms of this function of DDM1 remain unknown.
In this study, we focused on histone variant, H2A.W, which localizes at TE-rich heterochromatic regions. Our genomic analysis revealed that H2A.W was lost from heterochromatin when DDM1 is non-functional. DDM1 directly binds to H2A.W. Histone binding regions of DDM1 are important for H2A.W deposition and TE silencing. These results suggest that DDM1 silences TEs by deposition of H2A.W into heterochromatin. This pathway to silence TEs by chromatin remodeling could be conserved to mammals, since mammals have DDM1 and H2A.W orthologs with similar functions.
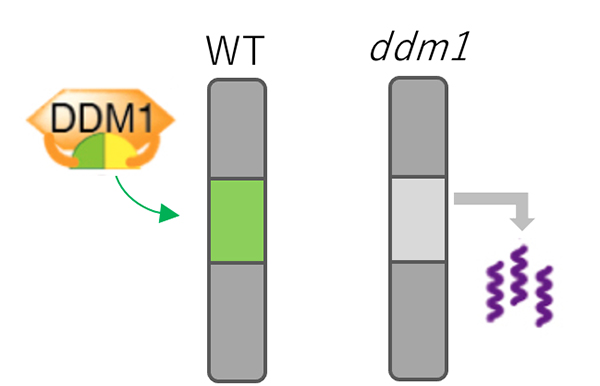
Figure: Mechanism for TE silencing regulated by DDM1.
In wild type plant (left), DDM1 mediates deposition of H2A.W (shown by green) into genomic regions with silent TEs. In ddm1 mutants (right), H2A.W was lost from heterochromatin, which is associated with derepression of TEs.










