データで見る遺伝学コース
年度別志願者・合格者・入学者数
| 年度 | 2024 | 2023 | 2022 | 2021 | 2020 | |||||
| 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | |
| 志願者 | 10 | 3 | 13 | 5 | 5 | 2 | 9 | 5 | 5 | 3 |
| 合格者 | 8 | 3 | 9 | 5 | 4 | 2 | 6 | 3 | 5 | 2 |
| 入学者 | 8 | 3 | 7 | 5 | 4 | 1 | 5 | 3 | 4 | 2 |
| 年度 | 2019 | 2018 | 2017 | 2016 | 2015 | |||||
| 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | |
| 志願者 | 7 | 6 | 7 | 3 | 4 | 1 | 11 | 1 | 7 | 2 |
| 合格者 | 4 | 2 | 6 | 3 | 2 | 1 | 6 | 0 | 3 | 1 |
| 入学者 | 4 | 2 | 5 | 3 | 2 | 1 | 6 | 0 | 3 | 1 |
奨学金(日本学生支援機構)の貸与状況
| 年度 | 2024 | 2023 | 2022 | 2021 | 2020 | |||||
| 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | |
| 応募者数 | 4 | 1 | 0 | 1 | 3 | 1 | 1 | 1 | 3 | 0 |
| 採用者数 | 4 | 1 | 0 | 1 | 3 | 1 | 1 | 1 | 3 | 0 |
| 年度 | 2019 | 2018 | 2017 | 2016 | 2015 | |||||
| 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | 5年一貫制 博士課程 | 博士 後期課程 | |
| 応募者数 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 2 |
| 採用者数 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 2 |
学術振興会特別研究員採用状況
| 年度 | 2024 | 2023 | 2022 | 2021 | 2020 | |||||
| DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | |
| 申請者数 | 1 | 5 | 5 | 3 | 4 | 9 | 3 | 3 | 3 | 4 |
| 採用者数 | 0 | 1 | 2 | 0 | 0 | 5 | 0 | 0 | 1 | 1 |
| 年度 | 2019 | 2018 | 2017 | 2016 | 2015 | |||||
| DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | DC1 | DC2 | |
| 申請者数 | 1 | 5 | 4 | 6 | 2 | 5 | 5 | 7 | 2 | 7 |
| 採用者数 | 0 | 1 | 0 | 3 | 1 | 2 | 0 | 4 | 1 | 3 |
学位取得状況
| 年度 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 |
| 課程博士 | 9 | 7 | 9 | 3 | 6 | 3 | 6 | 12 | 7 | 6 |
| 論文博士 | 1 | 0 | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
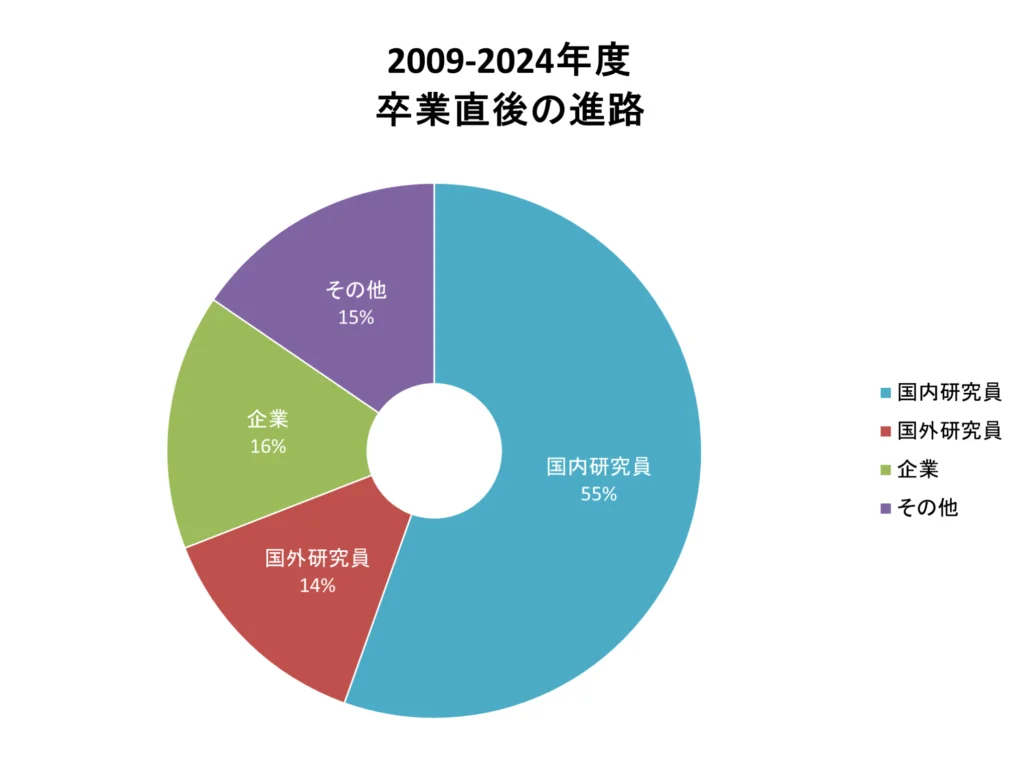
大学院修了後の進路・就職状況