



Press release
0 (preface).
Preface to the special issue on the Yaponesia Genome Project
Naruya Saitou
1 (original article).
Modern human DNA analyses with special reference to the Inner-Dual structure model of Yaponesian
Timothy Jinam, Yosuke Kawai, and Naruya Saitou
2 (original article).
Ancient genomes from the initial Jomon period: new insights into the genetic history of the Japanese archipelago
Noboru Adachi, Hideaki Kanzawa-Kiriyama, Nara T., Kakuda T., Nishida I., and Ken-ichi Shinoda
3 (original article).
The time-dependent evolutionary rate of mitochondrial DNA in small mammals inferred from biogeographic calibration points with reference to the late Quaternary environmental changes
Hitoshi Suzuki
4 (original article).
Geographical distribution of certain toponyms in the Samguk Sagi
Mitsuaki Endo
5 (review article).
Exploring models of human migration to the Japanese archipelago using genome-wide genetic data
Naoki Osada and Yosuke Kawai
6 (review article).
Paleogenomics of human remains in East Asia and Yaponesia focusing on current advances and future directions
Kae Koganebuchi and Hiroki Oota
Abstract of Paper 1:
Previous studies suggested two major migration events during the Jomon and Yayoi periods that affected the genetic diversity of modern Japanese (Yaponesians). We explored the possibility of a three-wave migration model by examining three datasets of modern human DNA: (1) whole mitochondrial (mt) DNA genomes of 1642 Yaponesians; (2) mtDNA haplogroup frequencies of 59105 Yaponesians from 47 prefectures; and (3) genome-wide SNP data of two Yaponesians (Ainu, Okinawa) and whole-genome sequence data of Yamato individuals, the Funadomari Jomon F23 individual, and three East Asian populations (Korean, northern Chinese, and southern Chinese). Past population size change was estimated based on dataset 1, and we clearly observed a steep population increase after the Yayoi period. Principal-component analysis and phylogenetic network analysis were applied to dataset 2, and we confirmed the pattern consistent with our model. An admixture program was used on dataset 3, and we found that the two- and three-layer migration models are both compatible with these SNP data. Taken together, these three datasets provide support for our three-wave, ‘inner dual-structure’ model.
Abstract of Paper 2:
Starting 16000 years ago, the Neolithic lifestyle known as the Jomon culture spread across the Japanese archipelago. Although extensively studied by archaeology and physical anthropology, little is known about the genetic characteristics of the Jomon people. Here, we report the entire mitogenome and partial nuclear genome of skeletal remains from the initial Jomon period that were excavated from the Higashimyo shell midden site at Saga City, Kyushu Island, Japan. This is the first genome analysis of the initial Jomon people of Kyushu Island. These results provide important data for understanding the temporal transition and regional differences of the Jomon people. The mitochondrial DNA and Y-chromosome haplogroups were similar to those found in the previously reported later Jomon people. Moreover, comparison of three nuclear genomes from the initial to final Jomon periods indicated genetic continuity throughout the Jomon period within the Japanese archipelago with no significant evidence of admixture. This indicates that the genetic differentiation found among the Jomon people was promoted by the progression of regionalization throughout the Jomon period. Further accumulation of high-quality Jomon genome data spanning a wide range of regions and ages will clarify both intimate regional and temporal differences of the Jomon people and details of their admixture history with rice farmers, as suggested by Jomon mitochondrial genome data. The results obtained from this study provide important information for further analysis.
Abstract of Paper 3:
Mitochondrial DNA (mtDNA) sequences have long been the most popular marker for assessing phylogenetic relationships and uncovering population dynamics. However, the mechanism of the nucleotide substitution rate of mtDNA remains unclear. While the evolutionary rate over tens of thousands of years is thought to be time dependent, the overall picture is not fully understood. This article presents recent achievements related to the time-dependent evolutionary rate of mtDNA in small rodents in the Japanese archipelago. The method focuses on rapid expansion events during the late Quaternary, during which there was a prolonged severe cold period and repeated abrupt warm periods, providing multiple calibration points. The global sea level fluctuation and migration to islands help to specify the calibration points. For calibration points at 11000, 15000, 53000, and 130000 years ago, the evolutionary rates were approximately 0.11, 0.11, 0.047, and 0.029 substitutions/site/million years, respectively, in the mitochondrial cytochrome b gene (Cytb). Applying the higher rate to assess the evolutionary history of the commensal house mouse (Mus musculus) and complete mitochondrial genome sequences (~16000 bp) allowed us to trace prehistoric human culture development based on millet and rice agriculture. The pattern of time-dependent evolutionary rates presented here is likely applicable to other small rodents. The Japanese archipelago is ideal for assessing evolutionary rates with biogeographic calibration points in the late Quaternary in species with multiple genetically distinct local populations.
Abstract of Paper 4:
Kim Busik’s Samguk Sagi 三国史記, History of the Three Kingdoms, dating from 1145 A.D., is renowned for including Japanese toponyms in the Korean peninsula and the north of Yalujiang district (modern Liaoning and Jilin Provinces). Kim’s work recorded the older, archaic toponyms before they were converted into sinicized (i.e. expressed with Chinese words) place names with two Chinese characters in 757 A.D. by the order of King Kyeongdeok. This paper maps specific words included in the place names of 783 locations for which the corresponding present places are evident. The following were words examined: (1) ‘river’ and its related words; (2) ‘valley’; (3) ‘mountain’ and ‘ridge’; and (4) ‘city’ and ‘burg’. Japonic-sourced toponyms are typically distributed in the central and northern areas of the Yalu River, primarily in the district of Koguryŏ; however, they go beyond these regions. The use of Chinese loanwords is noted in the southern area, where determining which language was spoken is difficult. In a town near Seoul, the stem of the toponym belongs to the Korean language, whereas the unit word belongs to the Japonic language. This usage may be attributed to bilingualism, whereby Korean-speaking inhabitants used their own language for the stem of the place name. Mongolic and/or Tungusic loanwords are also found. In some cases, determining the language origin of the current toponyms is difficult. Therefore, the minute geographical distribution of the origin languages is displayed word by word. These toponyms reflect the traces of indigenous languages and reveal that Japonic-speaking people still dwelled in the central area of the peninsula and in the northern area of the Yalu River at that period.
Abstract of Paper 5:
The origins of people in the Japanese archipelago are of long-standing interest among anthropologists, archeologists, linguists, and historians studying the history of Japan. While the ‘dual-structure’ model proposed by Hanihara in 1991 has been considered the primary working hypothesis for three decades, recent advances in DNA typing and sequencing technologies provide an unprecedented amount of present-day and ancient human nuclear genome data, which enable us to refine or extend the dual-structure model. In this review, we summarize recent genome sequencing efforts of present-day and ancient people in Asia, mostly focusing on East Asia, and we discuss the possible migration routes and admixture patterns of Japanese ancestors. We also report on a meta-analysis we performed by compiling publicly available datasets to clarify the genetic relationships of present-day and ancient Japanese populations with surrounding populations. Because the ancient genetic data from the Japanese archipelago have not yet been fully analyzed, we have to corroborate models of prehistoric human movement using not only new genetic data but also linguistic and archeological data to reconstruct a more comprehensive history of the Japanese people.
Abstract of Paper 6:
Ancient DNA analysis became paleogenomics once high-throughput sequencing technology was applied to ancient DNA sequencing. Paleogenomics based on whole-genome information from Neanderthals and Denisovans showed that small fragments of these genomes remain in the modern human genome, and corresponding studies of anatomical modern humans clarified the history of migration and expansion among Homo sapiens. Due to geographical and environmental conditions, paleogenomic studies have fallen behind in Eastern compared with Western Eurasia. Recently, however, various capture sequencing techniques, which can enrich ancient DNA, have been used in East Eurasia, and the field of paleogenomics has been further developed. This review briefly introduces the history of ancient DNA analysis leading to paleogenomics, outlines three sequencing stages (partial, draft, and complete genome sequencing) and capture methods, and discusses the necessity of high-quality sequencing for paleogenomes of Eastern Eurasia.
Chemokine ligand 28 (CCL28) negatively regulates trabecular bone mass by suppressing osteoblast and osteoclast activities
Rina Iwamoto, Takumi Takahashi, Kazuto Yoshimi, Yuji Imai, Tsuyoshi Koide, Miroku Hara, Tadashi Ninomiya, Hiroaki Nakamura, Kazutoshi Sayama, Akira Yukita
Journal of Bone and Mineral Metabolism 2021 March 15 DOI:10.1007/s00774-021-01210-9
Healthy adult bone mass is tightly controlled by regulator of bone formation and resorption (bone metabolism regulator). The disturbance of this regulation causes pathological bone loss, such as osteoporosis. Recently, the relationship between many chemokines and bone has not yet been investigated, although chemokine has been attracting attention as a bone metabolism regulator.
We performed experiments to clarify the role of CCL28 in bone metabolism, because CCR3 which receptor for CCL28, regulates bone metabolism.
Analysis of Ccl28 deficient mice bone tissue and cultured cell experiments revealed that CCL28 negatively regulates bone mass via suppresses the activation of osteoblasts and osteoclasts. Interestingly, we revealed that Ccl28 expression increased in aged mice bone tissue.
These results provide important insights into bone immunology and the selection of new osteoporosis treatments.
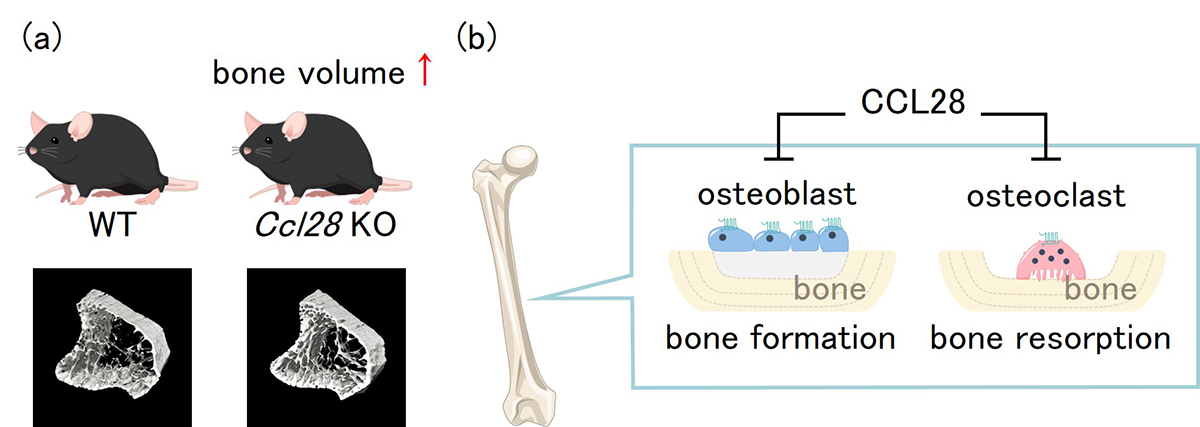
Figure: (a)μCT analysis of bone structure revealed that Ccl28 deficiency increased trabecular bone mass. (b)In vivo and In vitro experiments revealed that CCL28 negatively regulates bone mass by suppressing the activation of osteoblasts and osteoclasts in bone tissue.
Sycp1 Is Not Required for Subtelomeric DNA Double-Strand Breaks but Is Required for Homologous Alignment in Zebrafish Spermatocytes
Yukiko Imai, Kenji Saito, Kazumasa Takemoto, Fabien Velilla, Toshihiro Kawasaki, Kei-ichiro Ishiguro and Noriyoshi Sakai
Front Cell Dev Biol 9, 664377 (2021) DOI:10.3389/fcell.2021.664377
Meiosis is a specialized cell division, by which haploid gametes are produced from diploid cells for sexual reproduction. In meiosis, recombination mediates reciprocal exchanges of genetic contents between homologous chromosomes. In human and zebrafish males, meiotic recombination preferentially occurs near telomeres, but the mechanism underlaying the subtelomeric bias remains unknown.
In this study, we investigated roles of physical connection between homologous chromosomes (synapsis) in subtelomeric recombination by using zebrafish. We found that recombination begins near telomeres and homologous chromosomes pair locally, in synapsis-defective zebrafish. However, in the mutant zebrafish, a substantial proportion of pairing was lost in later stages, and no spermatozoa was generated. These observations revealed that synapsis is not essential for initiation of recombination at subtelomeres but is required for its completion.
Since the subtelomeric bias of recombination is also observed in human, new insights into zebrafish meiosis will facilitate understanding human infertility caused by meiotic defects.
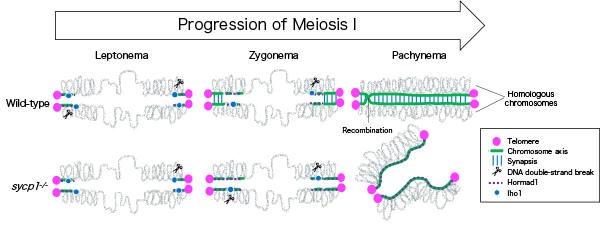
Figure: In wild-type zebrafish males, meiotic recombination begins with DNA double-strand breaks near telomeres. Synapsis is also initiated near telomeres at zygonema, and homologous chromosomes are paired up at pachynema. In sycp1 mutant males, DNA double-strand breaks and local and transient homologous pairing occur near telomeres.
Illuminating ALS Motor Neurons With Optogenetics in Zebrafish
Kazuhide Asakawa, Hiroshi Handa, Koichi Kawakami
Frontiers in Cell Developmental Biology 9, 640414 (2021) DOI:10.3389/fcell.2021.640414
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder characterized by progressive degeneration of motor neurons in the brain and spinal cord. Spinal motor neurons align along the spinal cord length within the vertebral column, and extend long axons to connect with skeletal muscles covering the body surface. Due to this anatomy, spinal motor neurons are among the most difficult cells to observe in vivo. Larval zebrafish have transparent bodies that allow non-invasive visualization of whole cells of single spinal motor neurons, from somas to the neuromuscular synapses. This unique feature, combined with its amenability to genome editing, pharmacology, and optogenetics, enables functional analyses of ALS-associated proteins in the spinal motor neurons in vivo with subcellular resolution. Here, we review the zebrafish skeletal neuromuscular system and the optical methods used to study it. We then introduce a recently developed optogenetic zebrafish ALS model that uses light illumination to control oligomerization, phase transition and aggregation of the ALS-associated DNA/RNA-binding protein called TDP-43. Finally, we will discuss how this disease-in-a-fish ALS model can help solve key questions about ALS pathogenesis and lead to new ALS therapeutics.
This review article is a collaborative work conducted by Tokyo Medical University and NIG, and supported by the SERIKA FUND and KAKENHI (JP19K06933 and JP20H05345).
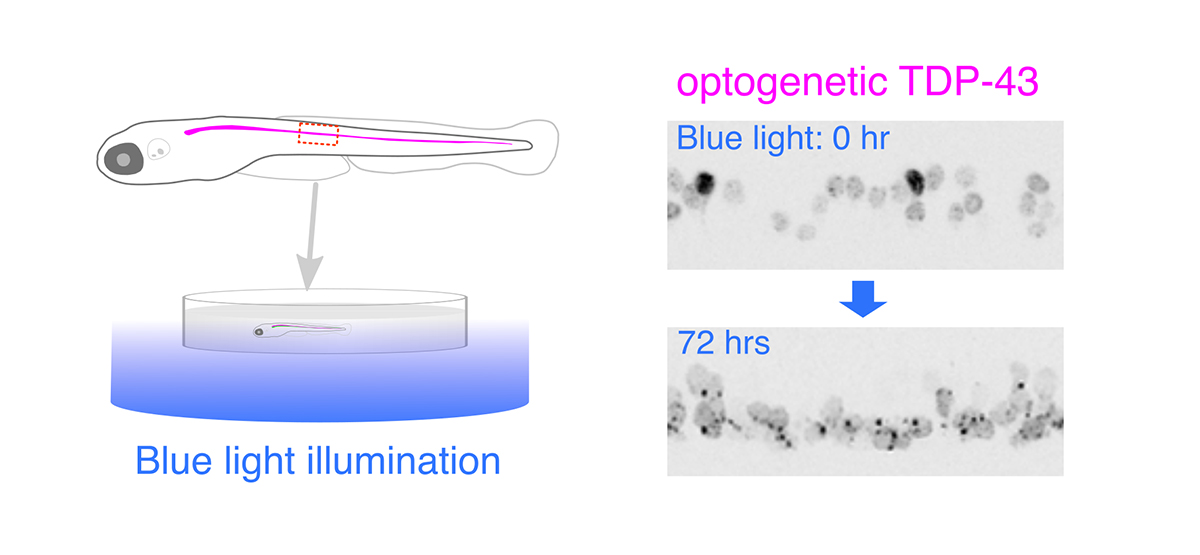
Figure: Optogenetic stimulation of TDP-43 phase transition in the zebrafish spinal motor neurons. Optogenetic TDP-43 forms abnormal aggregates by blue light illumination.
The service has been restored as of April 6, 13:00.
Physical nature of chromatin in the nucleus
Kazuhiro Maeshima, Shiori Iida, and Sachiko Tamura
Cold Spring Harbor Perspectives in Biology The Nucleus (2nd edition) 13, a040675 (2021) DOI:10.1101/cshperspect.a040675
The full text of the article is here.
Genomic information is encoded on long strands of DNA, which are folded into chromatin and stored in a tiny nucleus. Nuclear chromatin is a negatively charged polymer composed of DNA, histones, and various nonhistone proteins. Because of its highly charged nature, chromatin structure varies greatly depending on the surrounding environment (e.g., cations, molecular crowding, etc.). New technologies to capture chromatin in living cells have been developed over the past ten years. Our view on chromatin organization has drastically shifted from a regular and static one to a more variable and dynamic one. Chromatin forms numerous compact dynamic domains that act as functional units of the genome in higher eukaryotic cells and locally appears liquid-like. Chromatin dynamics can be modulated by several physical or geometrical factors in the cell including cations, chromatin proteins, and transcription machinery. This dynamic behavior can govern various genome functions by changing DNA accessibility. Based on new evidences from versatile genomics and advanced imaging studies, we discuss the physical nature of chromatin in the crowded nuclear environment and how it is regulated. This review article was published in Cold Spring Harbor Perspectives in Biology, also as a chapter of “The Nucleus (2nd edition)”.
This work was supported by JSPS and MEXT KAKENHI grants (20H05936), a Japan Science and Technology Agency CREST grant (JPMJCR15G2), the Takeda Science Foundation, and the Uehara Memorial Foundation.
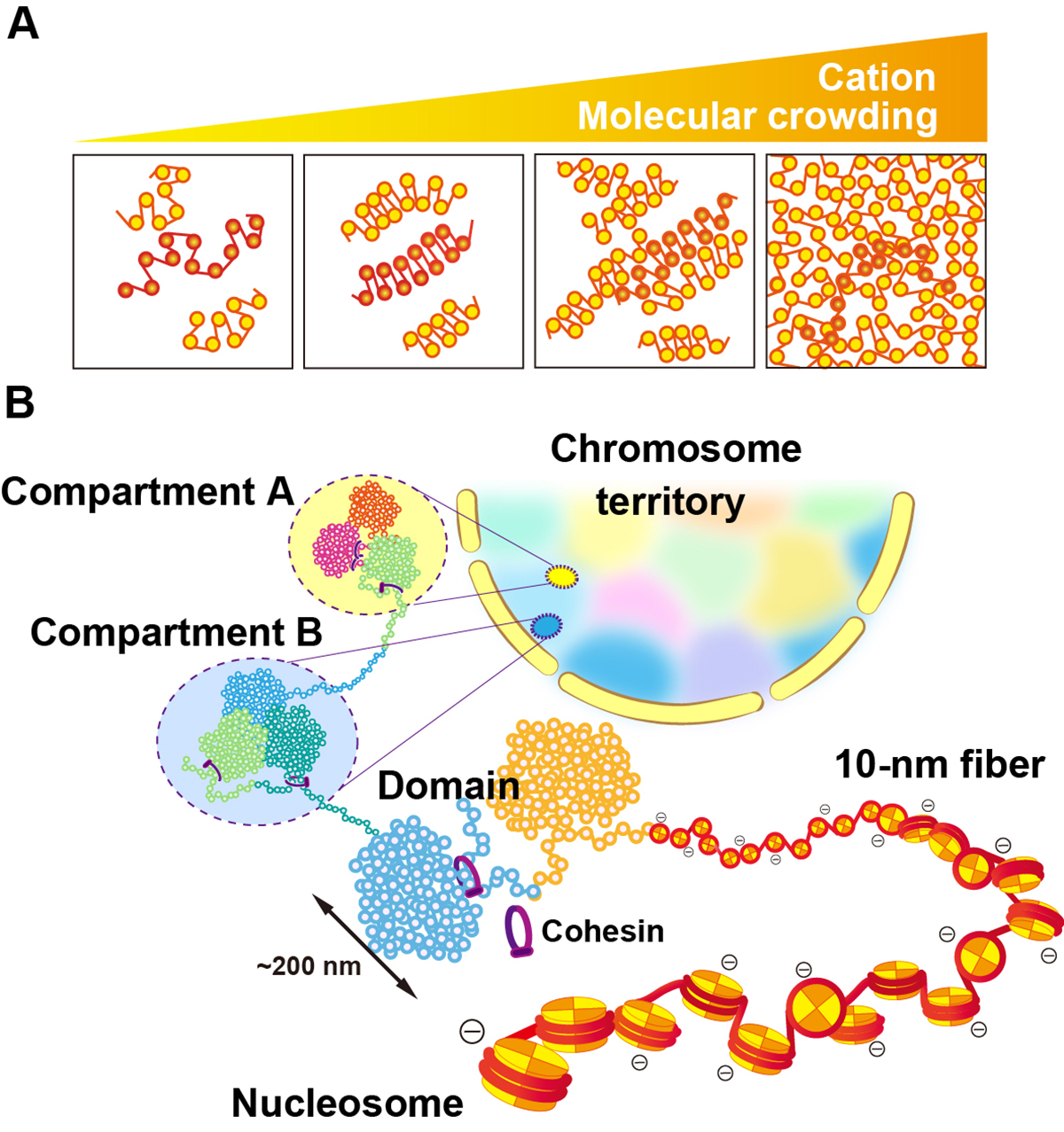
Figure: (A) Polymer melt model. Under physiological cation and/or molecular crowding conditions, inter-fiber nucleosomal contacts in the fibers increase and lead to a melted polymer-like or “sea of nucleosomes” state. (B) A simplified scheme for hierarchical chromatin organization in the nucleus: Nucleosome, the 10-nm fiber, chromatin domains, chromatin compartments and interphase chromosome occupied in a chromosome territory (highlighted as different colors).
Possible roles for the hominoid-specific DSCR4 gene in human cells
Morteza M. Saber, Marziyeh Karimiavargani, Takanori Uzawa, Nilmini Hettiarachchi, Michiaki Hamada, Yoshihiro Ito and Naruya Saitou
Genes and Genetic Systems 2021 March 24 DOI:10.1266/ggs.20-00012
00012 Down syndrome in humans is caused by trisomy of chromosome 21. DSCR4 (Down syndrome critical region 4) is a de novo-originated protein-coding gene present only in human chromosome 21 and its homologous chromosomes in apes. Despite being located in a medically critical genomic region and an abundance of evidence indicating its functionality, the roles of DSCR4 in human cells are unknown. We used a bioinformatic approach to infer the biological importance and cellular roles of this gene. Our analysis indicates that DSCR4 is likely involved in the regulation of interconnected biological pathways related to cell migration, coagulation and the immune system. We also showed that these predicted biological functions are consistent with tissue-specific expression of DSCR4 in migratory immune system leukocyte cells and neural crest cells (NCCs) that shape facial morphology in the human embryo. The immune system and NCCs are known to be affected in Down syndrome individuals, who suffer from DSCR4 misregulation, which further supports our findings. Providing evidence for the critical roles of DSCR4 in human cells, our findings establish the basis for further experimental investigations that will be necessary to confirm the roles of DSCR4 in the etiology of Down syndrome.
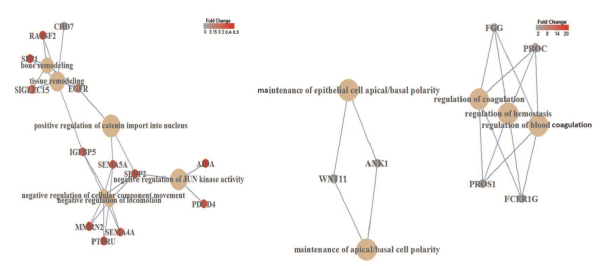
Figure: Gene regulatory network analysis of DSCR4 overexpression-mediated diffentially expressed genes. Left: diffentially expressed genes down-regulated by DSCR4 overexpression revealed enrichment of six interconnected pathways involved in the regulation of migration, locomotion and remodeling processes. Right: up-regulated diffentially expressed genes showed enrichment for three interconnected pathways involved in the regulation of blood coagulation and hemostasis along with maintenance of apical/basal cell polarity.
Dr. Shigehiro Kuraku joined NIG as a professor on April 1.
KURAKU, Shigehiro : Molecular Life History Laboratory

Dr. Hiroshi Mori opened a new laboratory as an associate professor on April 1.
MORI, Hiroshi Associate Professor : Advanced Genomics Center / Genome Diversity Laboratory

Two assistant professors joined NIG.
SAITO, Kei : Shimamoto Group • Physics and Cell Biology Laboratory

The TOYOTA Frontier Research Center has published an interview article introducing the joint research with Dr. Koichi Higashi, Assistant Prof. Hiroshi Mori, and Prof. Ken Kurokawa in the NIG Genome Evolution Laboratory. The article introduces “Biophilic ScoreTM” for measuring air quality in the environment.
| TOYOTA Website | |
| Kurokawa Group • Genome Evolution Laboratory |
Chromatin-based mechanisms to coordinate convergent overlapping transcription.
Soichi Inagaki, Mayumi Takahashi, Kazuya Takashima, Satoyo Oya, Tetsuji Kakutani.
Nature Plants 2021 March 1 DOI:10.1038/s41477-021-00868-3
In addition to coding genes, non-coding regions are often transcribed to make the transcription pattern overlapped. How organisms deal with overlapping transcription between neighboring genes remain unanswered. In this study we used a flowering plant Arabidopsis thaliana with a compact and gene-dense genome and found a mechanism to coordinate overlapping transcription in the opposite directions. We found that a histone demethylase protein FLD (flowering locus D) specifically controls genes with overlapping transcription by removing monomethylation of histone H3 lysine 4 (H3K4me1). We also showed that this regulation by FLD is linked to the mechanism to prevent the supercoiling or entanglement of DNA, which is crucial to maintain the genome. Interestingly, this global mechanism to coordinate overlapping transcription is utilized by plants to know the timing when they make flowers by adjusting the gene expression epigenetically in response to temperature. The findings of this study would lead to an understanding of the mechanism to cope with the crowded genome to properly regulate gene expression and respond to environment.
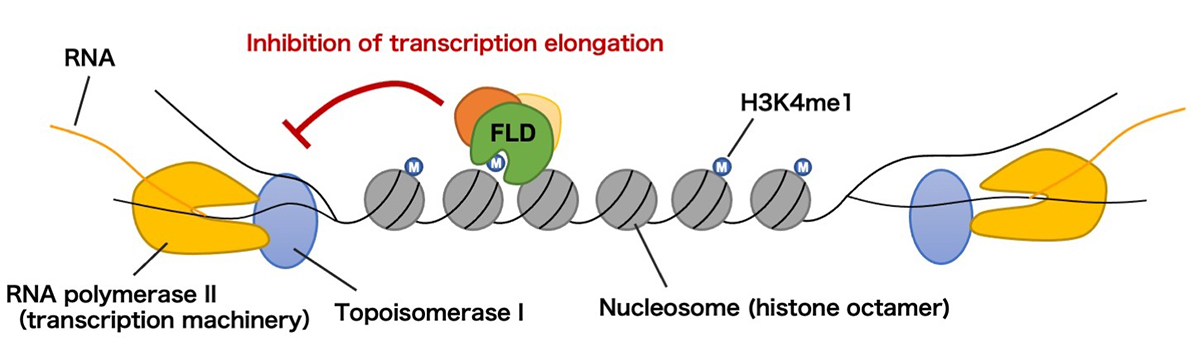
Figure: Proposed model of the findings.
RNA polymerase II transcribes DNA with opening the double helix. Topoisomerase I resolves DNA supercoils to assist transcription elongation. H3K4me1 accumulates in the transcribed regions of genes, but in cases where transcription takes place from both directions, FLD removes H3K4me1 to repress transcription elongation and coordinate bidirectional transcription.
Press release
Genome-wide SNP data of Izumo and Makurazaki populations support inner-dual structure model for origin of Yamato people
Timothy Jinam, Yosuke Kawai, Yoichiro Kamatani, Shunro Sonoda, Kanro Makisumi, Hideya Sameshima, Katsushi Tokunaga, and Naruya Saitou
Journal of Human Genetics 2021 January 25. DOI:10.1038/s10038-020-00898-3
Press release (In Japanese only)
The “Dual Structure” model on the formation of the modern Japanese population assumes that the indigenous hunter- gathering population (symbolized as Jomon people) admixed with rice-farming population (symbolized as Yayoi people) who migrated from the Asian continent after the Yayoi period started. The Jomon component remained high both in Ainu and Okinawa people who mainly reside in northern and southern Japan, respectively, while the Yayoi component is higher in the mainland Japanese (Yamato people). The model has been well supported by genetic data, but the Yamato population was mostly represented by people from Tokyo area. We generated new genome-wide SNP data using Japonica Array for 45 individuals in Izumo City of Shimane Prefecture and for 72 individuals in Makurazaki City of Kagoshima Prefecture in Southern Kyushu, and compared these data with those of other human populations in East Asia, including BioBank Japan data. Using principal component analysis, phylogenetic network, and f4 tests, we found that Izumo, Makurazaki, and Tohoku populations are slightly differentiated from Kanto (including Tokyo), Tokai, and Kinki regions. These results suggest the substructure within Mainland Japanese maybe caused by multiple migration events from the Asian continent following the Jomon period, and we propose a modified version of “Dual Structure” model called the “Inner-Dual Structure” model.
This study was supported by SOKENDAI Cooperative Research grant on modern human evolution, MEXT Grant-in-aid for Scientific Research on Innovative Areas “Yaponesian Genome” (grant number 18H05505), cooperative research of the Inter-University Research Institutions (grant number I- URIC18P01), and a cooperative research with Genesis Healthcare.
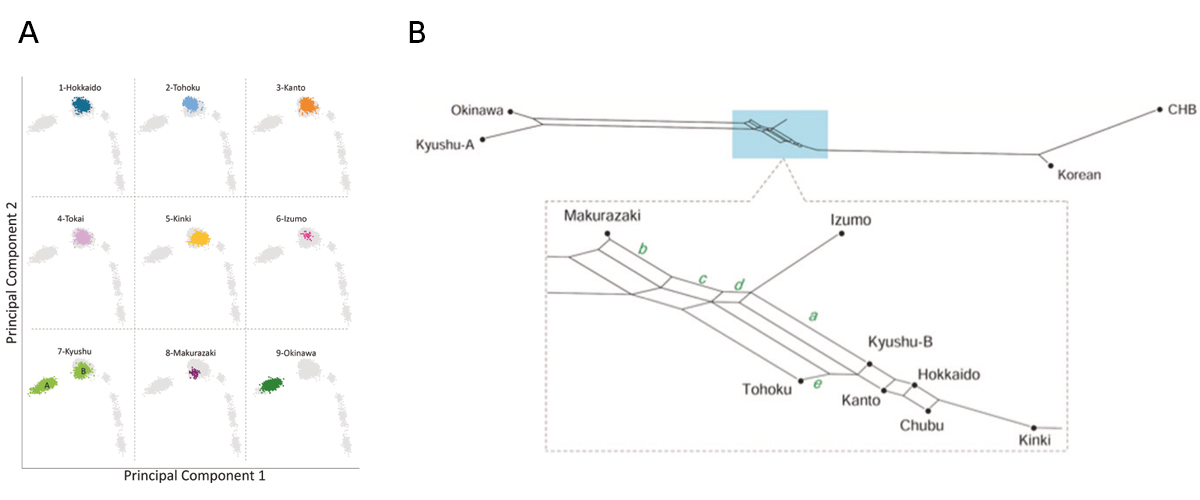
Figure: Genetic relationship of nine Yaponesian populations and some continental populations.
(A) Principal Component Analysis (PCA). DNA data for Izumo (6) and Makurazaki (8) are newly determined, and those for other seven regions are from BioBank Japan. Five continental populations are located at lower right side. Kyushu (7) population is divided into Okinawa cluster A and Yamato cluster B. (B) Phylogenetic network analysis using the Neighbor-Net method. Okinawa and Kyushu-A are located at left and Korean and CHB (Chinese Han in Beijing) are located at right. Makurazaki is the closest to the left cluster followed by Izumo, while Kinki is the closest to the right cluster.
Non-thalamic origin of zebrafish sensory nuclei implies convergent evolution of visual pathways in amniotes and teleosts
Solal Bloch, Hanako Hagio, Manon Thomas, Aure´ lie Heuze´, Jean-Michel Hermel, Elodie Lasserre, Ingrid Colin, Kimiko Saka, Pierre Affaticati, Arnim Jenett, Koichi Kawakami, Naoyuki Yamamoto, Kei Yamamoto
Elife 9, e54945 (2020) DOI:10.7554/eLife.54945
Ascending visual projections similar to the mammalian thalamocortical pathway are found in a wide range of vertebrate species, but their homology is debated. To get better insights into their evolutionary origin, we examined the developmental origin of a thalamic-like sensory structure of teleosts, the preglomerular complex (PG), focusing on the visual projection neurons. Similarly to the tectofugal thalamic nuclei in amniotes, the lateral nucleus of PG receives tectal information and projects to the pallium. However, our cell lineage study in zebrafish reveals that the majority of PG cells are derived from the midbrain, unlike the amniote thalamus. We also demonstrate that the PG projection neurons develop gradually until late juvenile stages. Our data suggest that teleost PG, as a whole, is not homologous to the amniote thalamus. Thus, the thalamocortical-like projections evolved from a non-forebrain cell population, which indicates a surprising degree of variation in the vertebrate sensory systems.
This study was conducted as collaboration with Dr. Kei Yamamoto at Universite´ Paris-Saclay and CNRS and Dr. Naoyuki Yamamoto at Graduate School of Bioagricultural Sciences, Nagoya University. This study was partially supported by NBRP and NBRP/Genome Information upgrading program from AMED and NIG-JOINT (2013-A15).
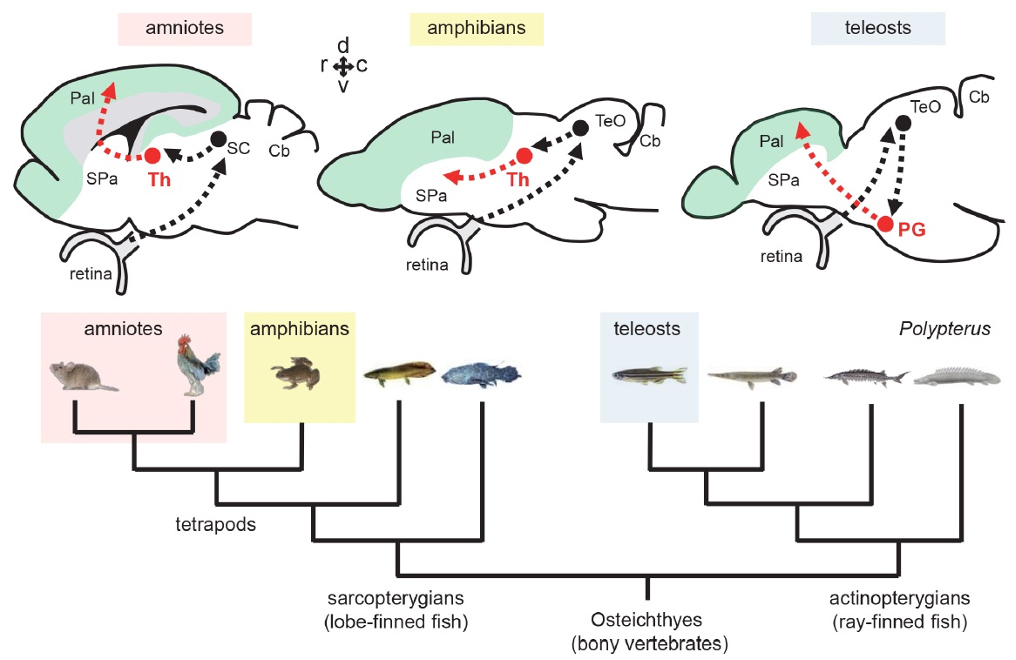
Figure1: Tectofugal pathways in amniotes, amphibians, and teleosts and their phylogenetic relationships.
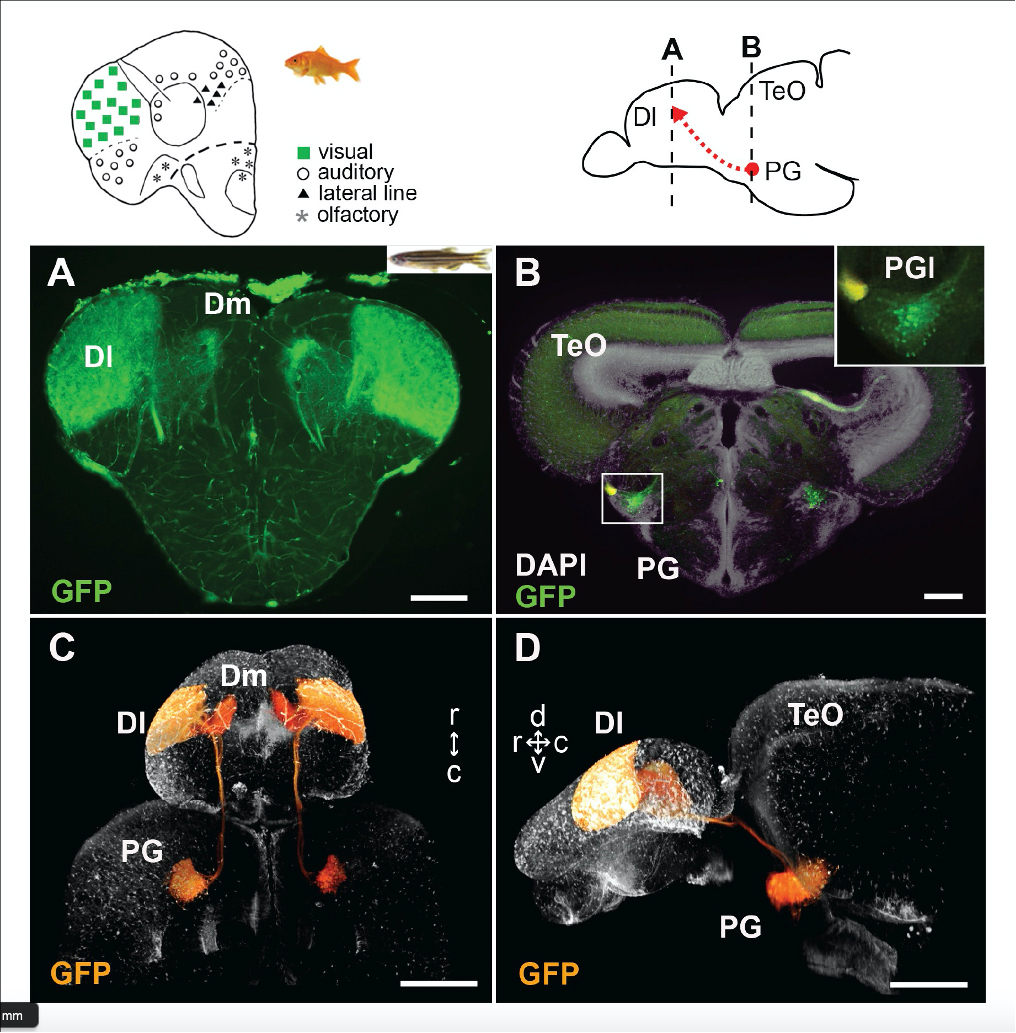
Figure2: Visualization of the preglomerular complex (PG) cells and their projection to the pallium in gene-trap transgenic fish.
1,6-hexanediol rapidly immobilizes and condenses chromatin in living human cells
Yuji Itoh, Shiori Iida, Sachiko Tamura, Ryosuke Nagashima, Kentaro Shiraki, Tatsuhiko Goto, Kayo Hibino, Satoru Ide and Kazuhiro Maeshima
Life Science Alliance 4, e202001005 (2021) DOI:10.26508/lsa.202001005
Liquid droplets formed inside the cell by liquid-liquid phase separation (LLPS) maintain membrane-less condensates/bodies (or compartments). These droplets are important for concentrating certain molecules and facilitating spatiotemporal regulation of cellular functions. 1,6-hexanediol (1,6-HD), an aliphatic alcohol, inhibits weak hydrophobic protein-protein/protein-RNA interactions required for the droplet formation (droplet melting activity) and is used here to elucidate the formation process of cytoplasmic/nuclear condensates/bodies. However, the effect of 1,6-HD on chromatin in living cells remains unclear. We found that 1,6-HD drastically suppresses chromatin motion and hyper-condenses chromatin in human cells by using live cell single-nucleosome imaging, which detects changes in the state of chromatin. These effects were enhanced in a dose-dependent manner. Chromatin was ‘frozen’ by 5%, or higher, concentrations of 1,6-HD. 1,6-HD greatly facilitated cation-dependent chromatin condensation in vitro. This 1,6-HD action is distinct from its melting activity of liquid droplets. Alcohols, such as 1,6-HD, appear to remove water molecules around chromatin and locally condense chromatin. Therefore, liquid droplet results obtained using 1,6-HD should be carefully interpreted or reconsidered when these droplets are associated with chromatin.
This work was supported by JSPS and MEXT KAKENHI grants (19K23735, 20J00572, 18K06187, 19H05273 and 20H05936), a Japan Science and Technology Agency CREST grant (JPMJCR15G2), Takeda Science Foundation, Uehara Memorial Foundation, NIG Postdoctoral Fellowship and JSPS Postdoctoral Fellowship (PD).
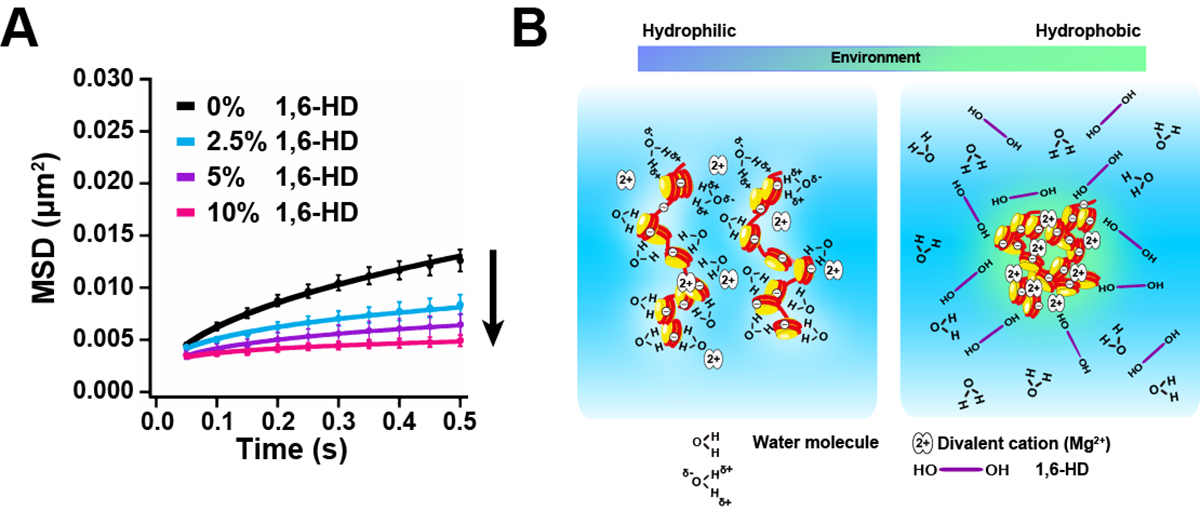
Figure: Chromatin immobilization by 1,6-HD and scheme for chromatin condensation.
(A) Mean square displacement plots (±SD among cells) of nucleosome motion in HeLa cells treated with 2.5% (light blue), 5% (purple), or 10% (pink) of 1,6-HD for 5–30 min. (B)(Left) Chromatin is associated with many water molecules with electrostatic interactions. (Right) Alcohols such as 1,6-HD can remove water molecules around chromatin, and its environment becomes more hydrophobic. This environmental change facilitates the formation of chromatin condensates. Note, this scheme is highly simplified and the molecules shown are not to scale. How 1,6-HD acts on chromatin at molecular level remains unclear.
The NIG international webinar by Prof. Kanemaki will be held on February 2nd at 4 pm, Eastern Time (on February 3rd at 6:00 am, Japan Standard Time). He will talk about a new genetic tool that enables rapid and reversible depletion of target proteins in living cells and animals. A Zoom link of the webinar will be obtained by free registration at the following URL (https://rois.zoom.us/meeting/register/tJAudOuprD4oHdV4dRlS6mAIdKX2F-jlAIEJ).
Time and Date:
Eastern Standard Time (EST): 4:00 pm, February 2nd
Pacific Standard Time (PST): 1:00 pm, February 2nd
Greenwich Mean Time (GMT): 9:00 pm, February 2nd
Central European Time (CET): 10:00 pm, February 2nd
Japan Standard Time (JST): 6:00 am, February 3rd
Registration (Zoom URL will be obtained by the free registration):
https://rois.zoom.us/meeting/register/tJAudOuprD4oHdV4dRlS6mAIdKX2F-jlAIEJ
Title:
AID2 enables rapid target protein degradation in living mammalian cells and mice
Speaker:
Prof. KANEMAKI, Masato
Molecular Cell Engineering Laboratory
National Institute of Genetics
Summary:
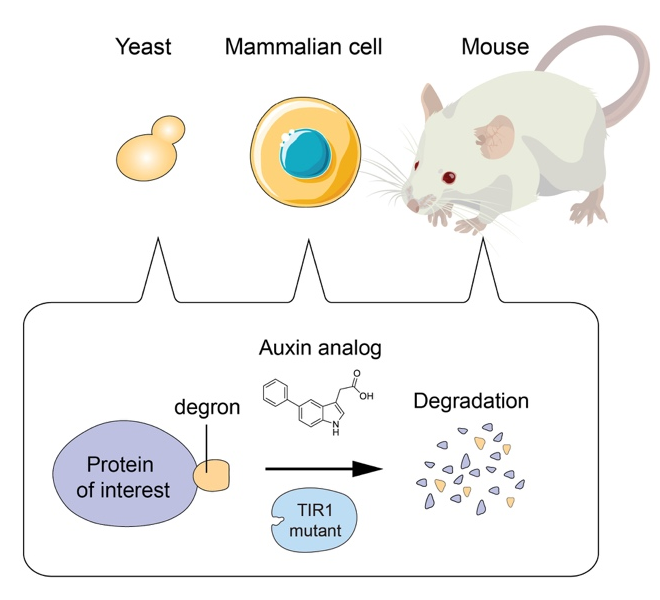
Genetic perturbation is a powerful way to analyze the function of proteins in living cells. For this purpose, we pioneered to develop the auxin-inducible degron (AID) technology by which a degron-fused protein can be rapidly degraded after the addition of the plant hormone auxin (Nishimura et al., Nat. Methods, 2009). By combining with CRISPR-based genome editing, it was possible to generate AID conditional mutants of human cells (Natsume et al., Cell Reports, 2016). The AID system became one of the popular genetic tools to study the function of proteins. However, leaky degradation and high doses of auxin for inducing degradation have been major drawbacks. Moreover, nobody has successfully applied the AID system to control protein degradation in living mice. We recently overcame these problems by taking advantage of chemical biology and successfully established the AID2 system (Yesbolatova et al. Nature Communications, 2020). By using AID2, we can now sharply control protein degradation in yeast, mammalian cells and mice.
• Link to KANEMAKI laboratory
• EurekAlert! link about the paper, Yesbolatova et al. Nature Communications, 2020
• Link to an interview of Prof. KANEMAKI
Chairperson: MAESHIMA, Kazuhiro










