



HLA-B*39:01:01 is a novel risk factor for antithyroid drug-induced agranulocytosis in Japanese population
Saya Nakakura, Kazuyoshi Hosomichi, Shinya Uchino, Akiko Murakami, Akira Oka, Ituro Inoue, Hirofumi Nakaoka
The Pharmacogenomics Journal 2020 September 22 DOI:10.1038/s41397-020-00187-4
Anti-thyroid drug (ATD) is a mainstay of Graves’ disease. About 0.1-0.5% of patients with Graves’ disease treated with ATD exhibit agranulocytosis, which is characterized by severe reduction of circulating neutrophils. Although it has been reported that the HLA class II allele (HLA-DRB1*08:03) was associated with ATD-induced agranulocytosis, the entire HLA region have not been explored in Japanese. Therefore, we performed HLA sequencing for 10 class I and 11 class II genes in 87 patients with ATD-induced agranulocytosis and 384 patients with Graves’ disease who did not develop ATD-induced agranulocytosis. By conducting case-control association studies at the HLA allele and haplotype levels, we identified HLA-B*39:01:01 as an independent risk factor. To verify the reproducibility of the association of HLA-B*39:01:01, we retrieved allele frequency data for HLA-B*39:01:01 from previous case-control association studies. The association of HLA-B*39:01:01 was significantly replicated in Chinese, Taiwanese, and European populations. A meta-analysis combining results from the previous and current studies reinforced evidence of association between HLA-B*39:01:01 and ATD-induced agranulocytosis. The results of this study will provide a better understanding of the pathogenesis of ATD-induced agranulocytosis in the context of HLA-mediated hypersensitivity reaction.
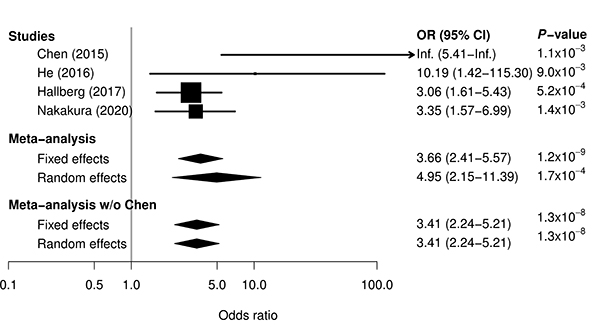
Figure: Meta-analysis for association between ATD-induced agranulocytosis and HLA-B*39:01:01 by combining previous three studies and current study. For single studies, odds ratio and 95% confidence interval are represented by box and whisker. For meta-analyses, odds ratio and 95% confidence interval are represented by a diamond. In all the single studies, HLA-B*39:01:01 was significantly associated with the risk for ATD-induced agranulocytosis. A meta-analysis combining results from the previous and current studies reinforced evidence of the association.
Developmental Phase Transitions in Spatial Organization of Spontaneous Activity in Postnatal Barrel Cortex Layer 4
Shingo Nakazawa, Yumiko Yoshimura, Masahiro Takagi, Hidenobu Mizuno, Takuji Iwasato.
Journal of Neuroscience 2020 September 4 DOI:10.1523/JNEUROSCI.1116-20.2020
Developing sensory cortices exhibit spatially-organized spontaneous activity, which is critical for the cortical circuit maturation. We previously reported “patchwork-type” spatial organization of spontaneous activity in the mouse barrel cortex at postnatal day 5 (P5) (Press release at 2018)
In the present study, we analyzed in detail how the spatial organization of barrel cortex spontaneous activity changes during the first two postnatal weeks. We found that spontaneous activity between P1 to P5 exhibited a patchwork-type pattern (Phase I). While around P9, a new type of pattern, showing wide area synchronization (Phase II), was observed, and at P11, neurons fired sparsely as observed in the adult brain (Phase III).
When thalamus was genetically silenced, Phase I cortical activity was abolished but Phase II and III activity remained intact, suggesting that the Phase I to II transition is associated with the shift of the activity source. On the other hand, the Phase II to III transition was impaired by cortical Rac1 inhibition. Phase II to III transition may be facilitated by Rac1-mediated synapse maturation.
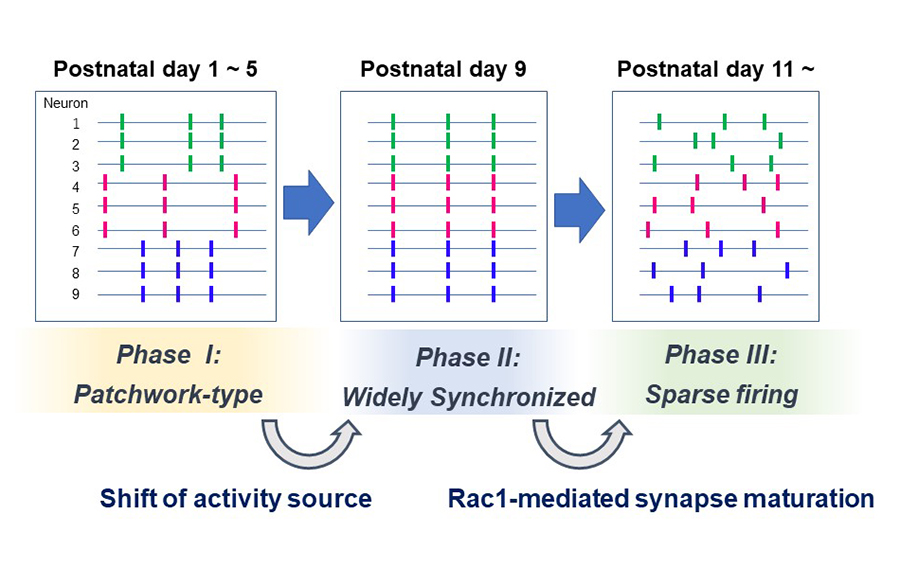
Figure: Three phases of spontaneous network activity were found in the barrel cortex layer 4 during the first two postnatal weeks. The Phase I to II transition is associated with the loss of thalamocortical input dependency. The Phase II to III transition may rely on cortical Rac1-dependent synapse maturation.
Zebrafish can regenerate endoskeleton in larval pectoral fin but the regenerative ability declines
Keigo Yoshida, Koichi Kawakami, Gembu Abe, Koji Tamura
Developmental Biology 463, 110-123 (2020). DOI:10.1016/j.ydbio.2020.04.010
We show for the first time endoskeletal regeneration in the developing pectoral fin of zebrafish. The developing pectoral fin contains an aggregation plate of differentiated chondrocytes (endochondral disc; primordium for endoskeletal components, proximal radials). The endochondral disc can be regenerated after amputation in the middle of the disc. The regenerated disc sufficiently forms endoskeletal patterns. Early in the process of regenerating the endochondral disc, epithelium with apical ectodermal ridge (AER) marker expression rapidly covers the amputation plane, and mesenchymal cells start to actively proliferate. Taken together with re-expression of a blastema marker gene, msxb, and other developmental genes, it is likely that regeneration of the endochondral disc recaptures fin development as epimorphic limb regeneration does. The ability of endoskeletal regeneration declines during larval growth, and adult zebrafish eventually lose the ability to regenerate endoskeletal components such that amputated endoskeletons become enlarged. Endoskeletal regeneration in the zebrafish pectoral fin will serve as a new model system for successful appendage regeneration in mammals.
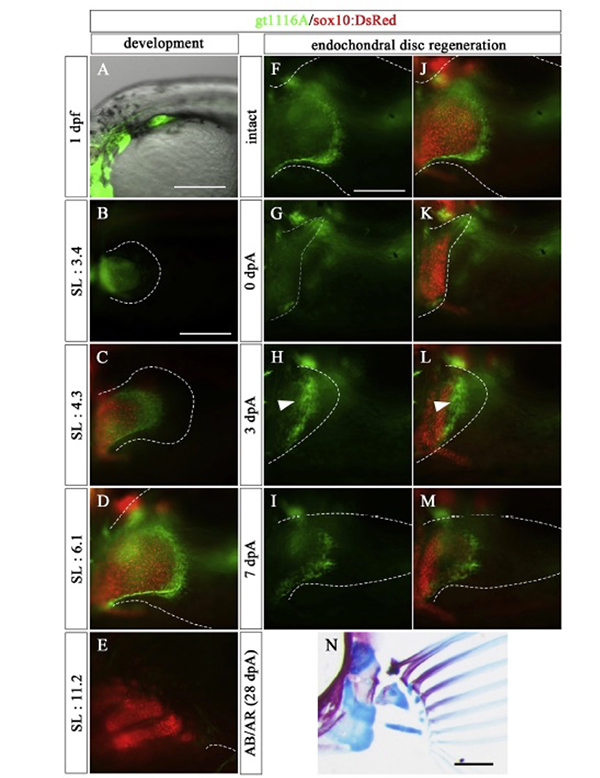
Figure: In the prdm16 gene trap line, the mesenchymal cells of the endoskeletal part of the pectoral fin expressed GFP (green) during the embryonic development. In this line, GFP was also expressed in the mesenchymal cells of the endoskeletal part during endochondrial disc regeneration. The red color in the figure indicates the expression of the sox10 gene, and labeled chondrocytes.
Press release
The dynamics, causes and impacts of mammalian evolutionary rates revealed by the analyses of capybara draft genome sequences
Isaac Adeyemi Babarinde and Naruya Saitou
Genome Biology and Evolution (2020) evaa157 DOI:10.1093/gbe/evaa157
Capybara (Hydrochoerus hydrochaeri) is the largest species among the extant rodents. The draft genome of capybara was sequenced with the estimated genome size of 2.6 Gbp. Although capybara is about 60 times larger than guinea pig, comparative analyses revealed that the neutral evolutionary rates of the two species were not substantially different. However, analyses of 39 mammalian genomes revealed very heterogeneous evolutionary rates. The highest evolutionary rate, 8.5 times higher than the human rate, was found in the Cricetidae-Muridae common ancestor after the divergence of Spalacidae. Muridae, the family with the highest number of species among mammals, emerged after the rate acceleration. Factors responsible for the evolutionary rate heterogeneity were investigated through correlations between the evolutionary rate and longevity, gestation length, litter frequency, litter size, body weight, generation interval, age at maturity, and taxonomic order. The regression analysis of these factors showed that the model with three factors (taxonomic order, generation interval and litter size) had the highest predictive power (R2 = 0. 74). These three factors determine the number of meiosis per unit time. We also conducted transcriptome analysis, and found that the evolutionary rate dynamics affects the evolution of gene expression patterns.

Left Panel: The memory of capybara “Rai-chan” (Photo: Izu Shaboten Zoo)
right Panel:Dr. Babarinde (from Nigeria, Africa), who received his Ph.D. from the Graduate University for Advanced Studies, SOKENDAI, in Saito’s lab and is currently a post-doctoral fellow in China.
Press release
Efficient open cultivation of cyanidialean red algae in acidified seawater
Shunsuke Hirooka, Reiko Tomita, Takayuki Fujiwara, Mio Ohnuma, Haruko Kuroiwa, Tsuneyoshi Kuroiwa and Shin-ya Miyagishima
Scientific Reports (2020)10: 13794 DOI:10.1038/s41598-020-70398-z
Press release (In Japanese only)
Microalgae possess high potential for producing pigments, antioxidants, and lipophilic compounds for industrial applications. However, their open pond cultures are often contaminated by other undesirable organisms, including their predators. In addition, the cost of using freshwater is relatively high, which limits the location and scale of cultivation compared with using seawater. It was previously shown that Cyanidium caldarium and Galdieria sulphuraria, but not Cyanidioschyzon merolae grew in media containing NaCl at a concentration equivalent to seawater. We found that the preculture of C. merolae in the presence of a moderate NaCl concentration enabled the cells to grow in the seawater-based medium. The cultivation of cyanidialean red algae in the seawater-based medium did not require additional pH buffering chemicals. In addition, the combination of seawater and acidic conditions reduced the risk of contamination by other organisms in the nonsterile open culture of C. merolae more efficiently than the acidic condition alone.
Source: S. Hirooka, et al., Scientific Reports (2020)10: 13794 DOI:10.1038/s41598-020-70398-z
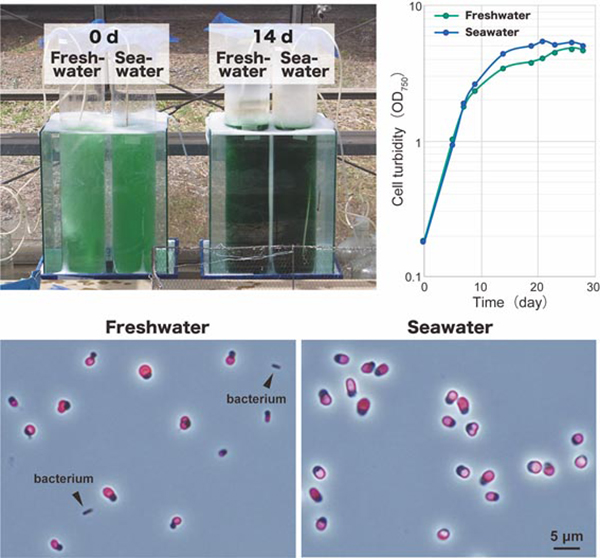
Fig: Outdoor cultivation of C. merolae in 7 L of nonsterile a freshwater-based medium and a seawater-based medium at day 0 and day 14. C. merolae cells in the seawater-based medium grew along a similar time course and to similar amounts compared with cells cultured in the freshwater-based medium. The microscopic observation of cultures 14 days after inoculation showed that the culture in the freshwater-based medium was contaminated with bacteria. In contrast, no bacteria or organisms other than C. merolae were observed in the culture in the seawater-based medium. Images were obtained by phase contrast microscopy, and the fluorescence images of chloroplasts (red) were overlaid.
Relationship between cell cycle and diel transcriptomic changes in metabolism in a unicellular red alga.
Takayuki Fujiwara, Shunsuke Hirooka, Ryudo Ohbayashi, Ryo Onuma, and Shin-ya Miyagishima.
Plant Physiology (2020) 183: 1484–1501 DOI:10.1104/pp.20.00469
Metabolism, cell cycle stages, and related transcriptomes in eukaryotic algae change with the diel cycle of light availability. In the unicellular red alga Cyanidioschyzon merolae, the S and M phases occur at night. To examine how diel transcriptomic changes in metabolic pathways are related to the cell cycle and to identify all genes, for which mRNA levels change depending on the cell cycle, we examined diel transcriptomic changes in C. merolae. In addition, we compared transcriptomic changes between the wild type and transgenic lines, in which the cell cycle was uncoupled from the diel cycle by the depletion of either cyclin-dependent kinase A (CDKA) or retinoblastoma-related (RBR) protein. Of 4,775 nucleus-encoded genes, the mRNA levels of 1,979 genes exhibited diel transcriptomic changes in the wild type. Of these, the periodic expression patterns of 454 genes were abolished in the transgenic lines, suggesting that the expression of these genes is dependent on cell cycle progression. The periodic expression patterns of most metabolic genes, except those involved in starch degradation and de novo dNTP synthesis, were not affected in the transgenic lines, indicating that the cell cycle and transcriptomic changes in most metabolic pathways are independent of the diel cycle. Approximately 40% of the cell–cycle–dependent genes were of unknown function, and approximately 19% of these genes of unknown function are shared with the green alga Chlamydomonas reinhardtii. The dataset presented in this study will facilitate further studies on the cell cycle and its relationship with metabolism in eukaryotic algae.
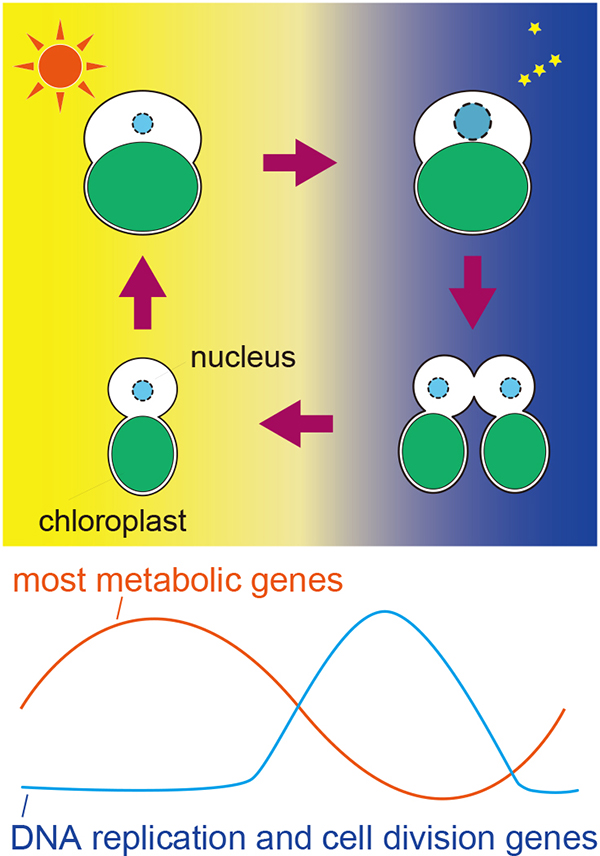
Figure: Cell growth and division of eukaryotic algae in accord with the diel cycle. Eukaryotic algae grow by photosynthesis in the daytime and undergo DNA replication and cell division in the night. Most metabolic genes are upregulated in the daytime whereas genes involved in DNA replication and cell division are induced dependently on cell cycle in the night.
NIG will be closed from August 14 to August 17, 2020 for summer holiday.
Thank you for your understanding and cooperation.
Cytoplasmic streaming drifts the polarity cue and enables posteriorization of the Caenorhabditis elegans zygote at the side opposite of sperm entry
Kenji Kimura and Akatsuki Kimura
Molecular Biology of the Cell (2020) 31: 1765–1773 DOI:10.1091/mbc.E20-01-0058
*FOURTH SPECIAL ISSUE on FORCES ON AND WITHIN CELLS
Cell polarization is required to define body axes during development. The position of spatial cues for polarization is critical to direct the body axes. In Caenorhabditis elegans zygotes, the sperm-derived pronucleus/centrosome complex (SPCC) serves as the spatial cue to specify the anterior-posterior axis. Approximately 30 minutes after fertilization, the contractility of the cell cortex is relaxed near the SPCC, which is the earliest sign of polarization and called symmetry breaking (SB). It is unclear how the position of SPCC at SB is determined after fertilization. Here, we show that SPCC drifts dynamically through the cell-wide flow of the cytoplasm, called meiotic cytoplasmic streaming (Figures A and B). This flow occasionally brings SPCC to the opposite side of the sperm entry site before SB (Figure C). Our results demonstrate that cytoplasmic flow determines stochastically the position of the spatial cue of the body axis, even in an organism like C. elegans for which development is stereotyped (Figure D).
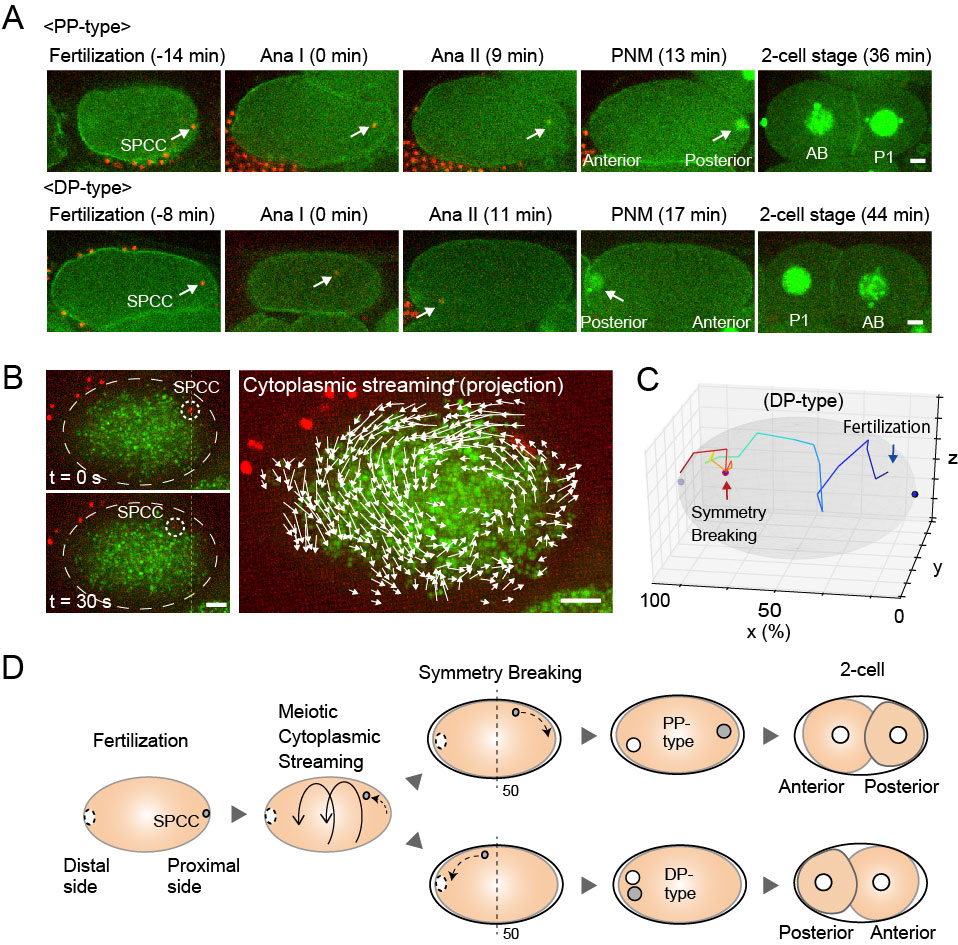
Figure: (A) Time series of the SPCC dynamics in C. elegans zygotes (white arrows). (B) SPCC movement (white dotted circle) correlates with the direction of cytoplasmic streaming (white arrows). (C) A trajectory of SPCC in DP-type. (D) Summary of the SPCC dynamics during cell polarization. Scale bars; 5 μm.
Kawakami Group / Laboratory of Molecular and Developmental Biology
Comparative Genomics Laboratory
NOGUCHI, Hideki / Center for Genome Informatics(CGI) / Advanced Genomics Center
The Genetic Basis of Morphological Diversity in Domesticated Goldfish
Tetsuo Kon, Yoshihiro Omori, Kentaro Fukuta, Hironori Wada, Masakatsu Watanabe, Zelin Chen, Miki Iwasaki, Tappei Mishina, Shin-ichiro S. Matsuzaki, Daiki Yoshihara, Jumpei Arakawa, Koichi Kawakami, Atsushi Toyoda, Shawn M. Burgess, Hideki Noguchi, Takahisa Furukawa.
Current Biology 30, 1-15 (2020). DOI:10.1016/j.cub.2020.04.034
Although domesticated goldfish strains exhibit highly diversified phenotypes in morphology, the genetic basis underlying these phenotypes is poorly understood. Here, based on analysis of transposable elements in the allotetraploid goldfish genome, we found that its two subgenomes have evolved asymmetrically since a whole-genome duplication event in the ancestor of goldfish and common carp. We conducted whole- genome sequencing of 27 domesticated goldfish strains and wild goldfish. We identified more than 60 million genetic variations and established a population genetic structure of major goldfish strains. Genome-wide association studies and analysis of strain-specific variants revealed genetic loci associated with several goldfish phenotypes, including dorsal fin loss, long-tail, telescope-eye, albinism, and heart-shaped tail. Our results suggest that accumulated mutations in the asymmetrically evolved subgenomes led to generation of diverse phenotypes in the goldfish domestication history. This study is a key resource for understanding the genetic basis of phenotypic diversity among goldfish strains.
This work was carried out as collaboration between Osaka Univ and NIG (Lab of Molecular and Developmental Biology, Comparative Genomics Lab and Advanced Genomics Center) and supported by the NIG-JOINT program

Figure: Wild type (1) and mutant (2-28) goldfish. Genome analysis revealed China (2-11), Ranchu (12-17) and Edo (18-28) groups.
RIF1 Controls Replication Initiation and Homologous Recombination Repair in a Radiation Dose-Dependent Manner
Yuichiro Saito, Junya Kobayashi, Masato T. Kanemaki, Kenshi Komatsu
Journal of Cell Science 2020 May 20. DOI:10.1242/jcs.240036
Genomic DNA is challenged by various types of stresses. Ionizing radiation (IR) cut DNA, generating a DNA damage called a DNA double-strand break (DSB). DSB is repaired via two pathways; an error-prone, ‘non-homologous end joining (NHEJ) pathway’ or a precise, ‘homologous recombination repair (HRR) pathway’. To maintain genome integrity, it is important for cells to choose an appropriate pathway for repairing DSBs.
It is believed that human cells dominantly use error-prone NHEJ to repair DSB. This idea came from the observations using high doses of IR. However, it has not been known whether the predominant use of NHEJ is applicable to DSBs induced by a low dose of IR. To answer this question, we used a low or high dose of IR and monitored the repair pathway. We found that HRR played a prominent role to repair DSBs after a low dose of IR, and it was suppressed after a high dose of IR. These results show that human cells sense radiation dose and choose an appropriate repair pathway. Furthermore, we identified RIF1 as a critical factor that controls HRR activity in an IR-dose dependent manner. We expect that these results would provide basic understandings of how radiation kills cancer cells in radiation therapy.
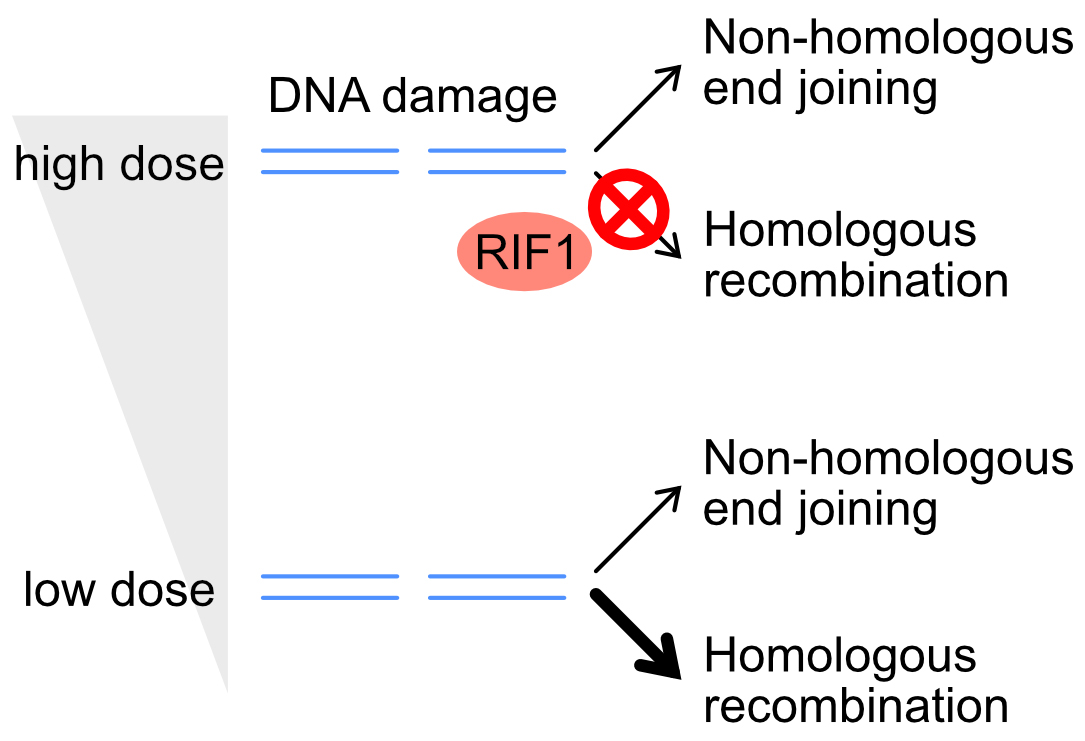
Figure: DNA double-strand break is repaired via the non-homologous end joining (NHEJ) or homologous recombination repair (HRR) pathway. RIF1 suppresses HRR only after a high dose of IR, directing cells towards using the NHEJ pathway.
Changes in the transcriptome, ploidy, and optimal light intensity of a cryptomonad upon integration into a kleptoplastic dinoflagellate
Ryo Onuma, Shunsuke Hirooka, Yu Kanesaki, Takayuki Fujiwara, Hirofumi Yoshikawa, Shin-ya Miyagishima
The ISME Journal (2020) DOI:10.1038/s41396-020-0693-4
Link to “Behind the Paper” the Nature Research Microbiology Community
Endosymbiosis of unicellular eukaryotic algae into previously nonphotosynthetic eukaryotes has established chloroplasts in several eukaryotic lineages. Additionally, certain unicellular organisms in several different lineages ingest algae and utilize them as temporal chloroplasts (kleptoplasts) for weeks to months before digesting them. Among these organisms, the dinoflagellate Nusuttodinium aeruginosum ingests the cryptomonad Chroomonas sp. and enlarges the kleptoplast with the aid of the cryptomonad nucleus. To understand how the cryptomonad nucleus is remodeled in the dinoflagellate, here we examined changes in the transcriptome and ploidy of the ingested nucleus. We show that, after ingestion, genes involved in metabolism, translation, and DNA replication are upregulated while those involved in sensory systems and cell motility are downregulated. In the dinoflagellate cell, the cryptomonad nucleus undergoes polyploidization that correlates with an increase in the mRNA levels of upregulated genes. In addition, the ingested nucleus almost loses transcriptional responses to light. Because polyploidization and loss of transcriptional regulation are also known to have occurred during the establishment of endosymbiotic organelles, these changes are probably a common trend in endosymbiotic evolution. Furthermore, we show that the kleptoplast and dinoflagellate are more susceptible to high light than the free-living cryptomonad but that the ingested nucleus reduces this damage.
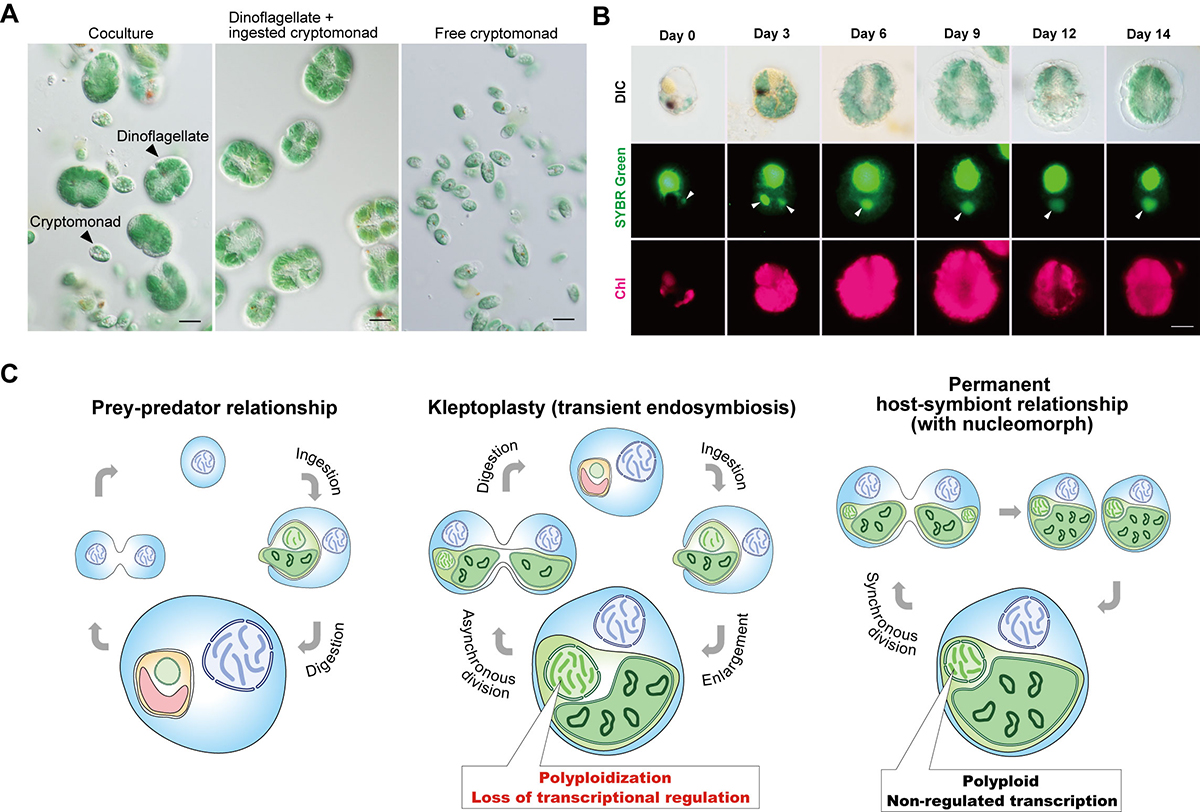
Figure: Kleptoplasty in Nusuttodinium aeruginosum.
(A) Micrographs showing the coculture of N. aeruginosum cells with Chroomonas sp.-derived kleptoplasts and free Chroomonas sp. (left). From the coculture, N. aeruginosum cells with the kleptoplasts (middle) were separated from free Chroomonas sp. (right) by filtration. Scale bar = 10 μm.
(B) Micrographs showing the change in the nucleus and kleptoplast derived from Chroomonas sp. in N. aeruginosum cells. Cells were stained with SYBR Green. Images of differential interference contrast (DIC), SYBR Green staining, and kleptoplast red fluorescence (Chl) are shown. The arrowhead indicates a nucleus derived from Chroomonas sp. The cell in the image from day 3 ingested two nuclei. Scale bar = 10 μm.
(C) Schematic comparison of prey–predator, transient endosymbiotic (kleptoplasty), and permanent endosymbiotic relationships. Predators start digesting algae immediately after phagocytic ingestion of algae (left). Kleptoplastic species retain the ingested algae for days to weeks in the cell before digesting them in some cases, including that of N. aeruginosum, with the nucleus of the algae (middle). The present study shows that the ingested nucleus is polyploidized in the host cell, which increases mRNA levels. The ingested nucleus almost loses transcriptional regulation. These changes were also common to the course of establishment of nucleomorph and chloroplast (also other types of plastids) as obligate endosymbionts in eukaryotes (right).
Press release
AnAms1.0: A high-quality chromosome-scale assembly of a domestic cat Felis catus of American Shorthair breed
Sachiko Isobe, Yuki Matsumoto, Claire Chung, Mika Sakamoto, Ting-Fung Chan, Hideki Hirakawa, Genki Ishihara, Hon-Ming Lam, Shinobu Nakayama, Shigemi Sasamoto, Yasuhiro Tanizawa, Akiko Watanabe, Kei Watanabe, Masaru Yagura, Yasukazu Nakamura
BioRχiv Posted May 19, 2020. DOI:10.1101/2020.05.19.103788
Press release (In Japanese only)
Researchers from the School of Life Sciences at The Chinese University of Hong Kong (CUHK) and Japanese research teams have constructed a high-resolution chromosome-scale full genome sequence assembly of an American Shorthair domestic cat (AnAms1.0). By incorporating data from multiple advanced genomic technologies, this genome assembly has a much improved quality over the currently available reference. This research will drive forward precision veterinary medicine to provide the most suitable treatments based on individual differences predicted from genomic information. Read More>
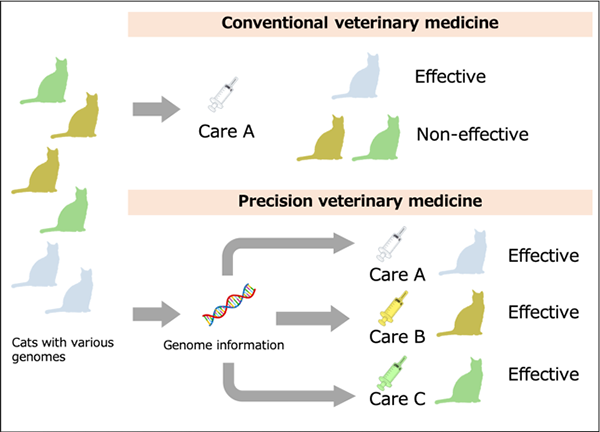
Fig: Personal Medicine for the Cat
In traditional veterinary medicine, the same treatment is given to any individual, where there exist cats not responsive to certain treatments. With the genomic approach, individually suited treatments can be applied based on individual traits predicted from genome information for more effective and efficient veterinary medicine.
Fluid-like chromatin: Toward understanding the real chromatin organization present in the cell
Kazuhiro Maeshima, Sachiko Tamura, Jeffrey C. Hansen and Yuji Itoh
Current Opinion in Cell Biology (2020) 64, 77-89. DOI:10.1016/j.ceb.2020.02.016
Eukaryotic chromatin is a negatively charged polymer consisting of genomic DNA, histones, and various non-histone proteins. Because of its highly charged character, the structure of chromatin varies greatly depending on the surrounding environment (i.e. cations etc.): from an extended 10-nm fiber, to a folded 30-nm fiber, to chromatin condensates/liquid-droplets. Over the last ten years, newly developed technologies have drastically shifted our view on chromatin from a static regular structure to a more irregular and dynamic one, locally like a fluid. Since no single imaging (or genomics) method can tell us everything and beautiful images (or models) can fool our minds, comprehensive analyses based on many technical approaches are important to capture actual chromatin organization inside the cell. Here we critically discuss our current view on chromatin and methodology used to support the view.
This research was supported by JST CREST (JPMJCR15G2), JSPS Kakenhi (19K23735, 16H04746, 16H06279 (PAGS) and 19H05273), Takeda Science Foundation, National Science Foundation grant (1814012), NIG Postdoctoral Fellowship and JSPS Postdoctoral Fellowship (PD).
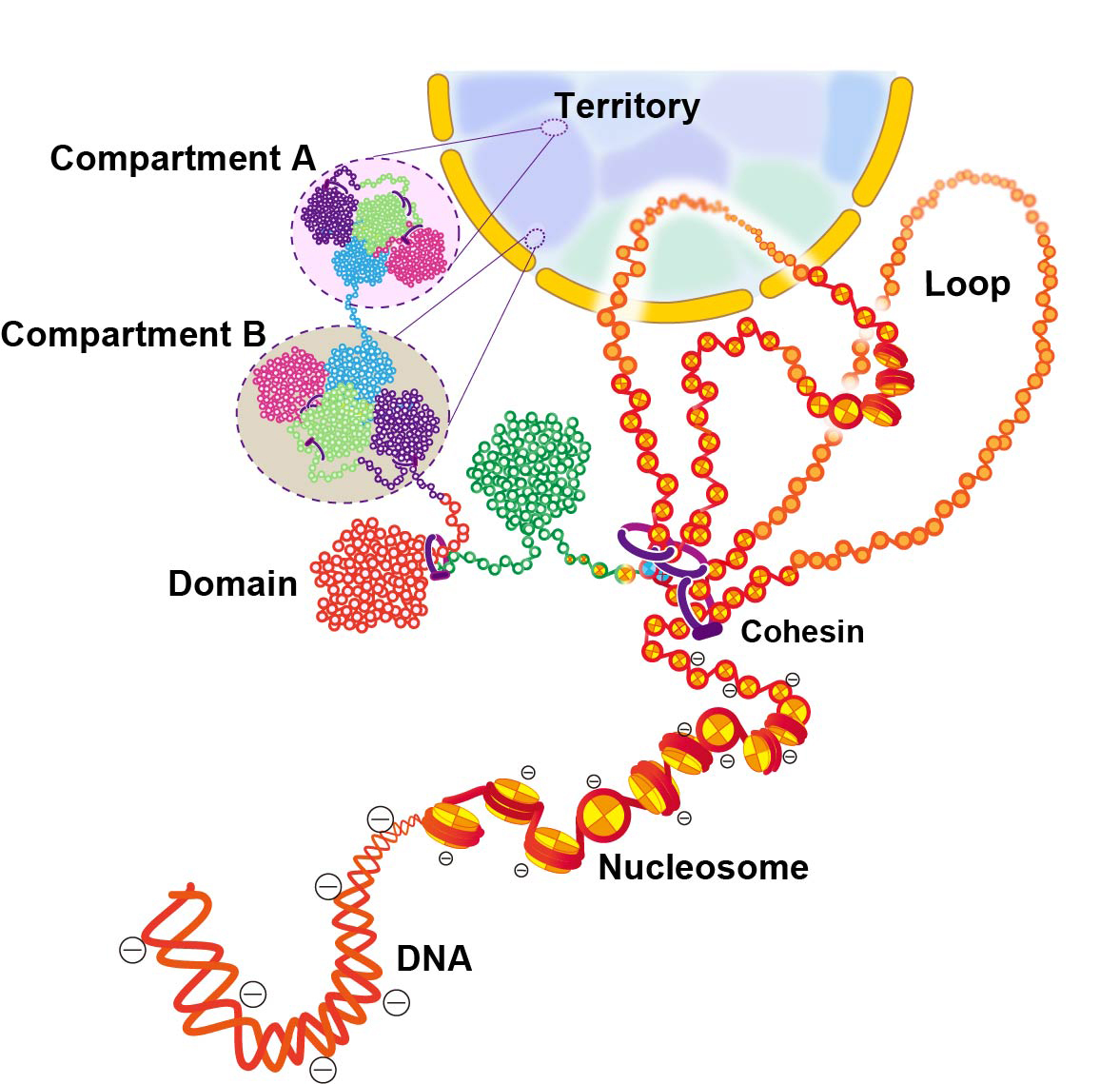
Figure: Scheme for hierarchical chromatin organization inside the cell nucleus.
DNA is wrapped around histone octamer to make a nucleosome. The 10-nm chromatin fiber can form a loop structure held by cohesin or other proteins. The chain of nucleosomes is compacted into chromatin domains, which interact over long distances to form chromatin compartments. A and B compartments in general represent transcriptionally active or open chromatin state (compartment A) and inactive or closed chromatin (compartment B), respectively. A single interphase chromosome consists of several compartments occupies that collectively form a chromosome territory. Note that this scheme is highly simplified and a more complex organization can be possible inside the cell.










