



Kimura Group • Cell Architecture Laboratory
Active fluctuations of cytoplasmic actomyosin networks facilitate dynein-driven transport
Takayuki Torisawa, Kei Saito, Ken’ya Furuta, Akatsuki Kimura
iScience (2025) Volume 28, Issue 12 114096 DOI:10.1016/j.isci.2025.114096
Inside cells, molecular motors transport cargo through a highly crowded cytoplasmic environment. While such an environment is assumed to hinder transport, its precise effect remains unclear. Here, we investigated how the dynamics of cytoplasmic environments affect dynein-driven transport in C. elegans early embryos. In living embryos, we found that an artificial dynein-cargo complex exhibited significantly faster transport than in vitro, indicating an active acceleration mechanism in vivo. By altering the activity of actomyosin networks, we found that dynein-driven transport was accelerated by actomyosin-driven cytoplasmic fluctuations, with speed increasing upon myosin upregulation and decreasing upon its depletion. Furthermore, in vitro force measurements of dynein suggest that the asymmetric force response to random forces, generated by fluctuating dynamics of actomyosin networks, may contribute to acceleration. This study provides insights into a regulatory mechanism of molecular motors within fluctuating cytoplasm, harnessing cytoplasmic fluctuations to enhance transport efficiency in a highly crowded environment.
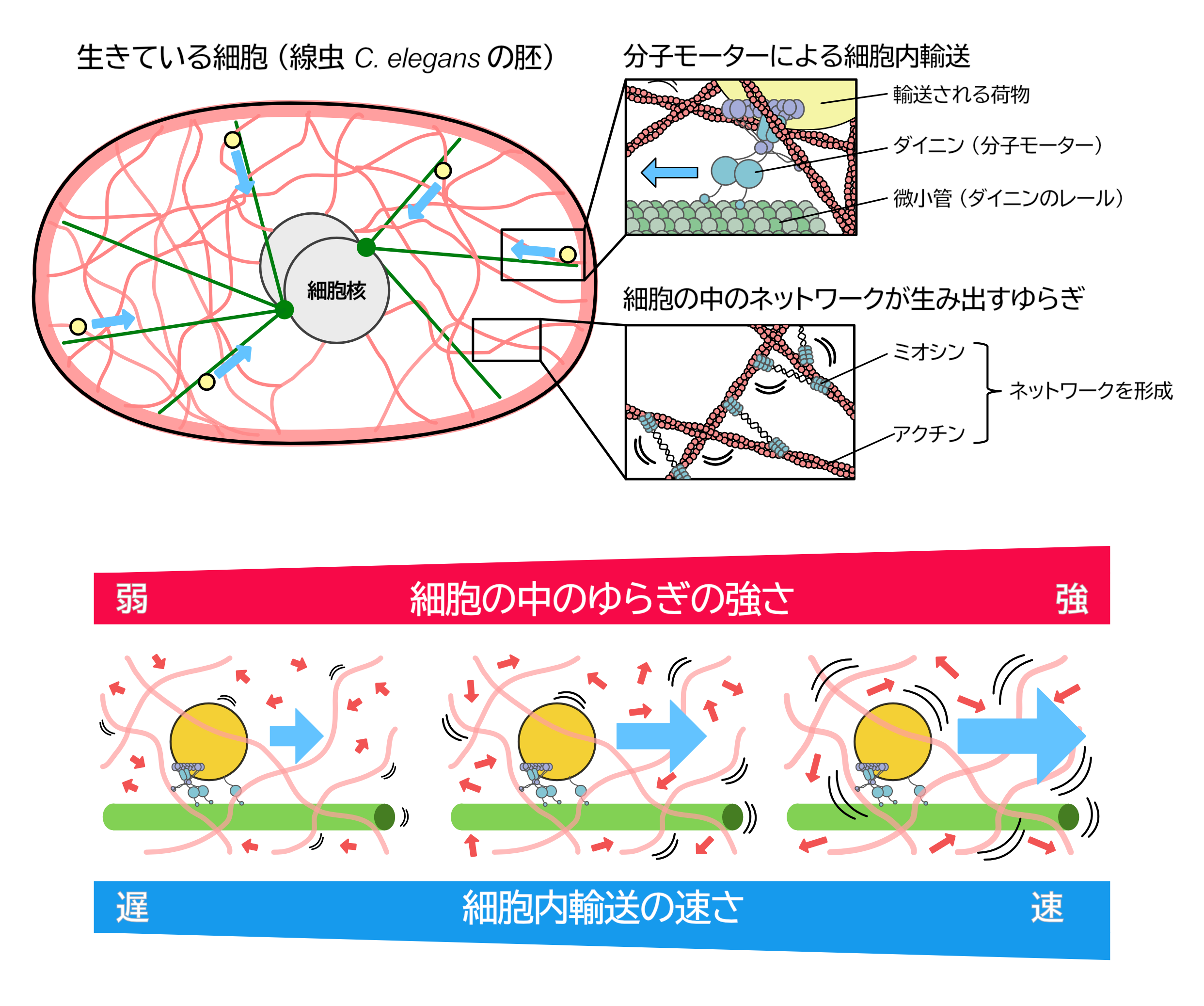
Figure: Model summarizing the biphasic change in local chromatin dynamics during doxycycline-induced transformation of EMR cells. The blue curve denotes a transient rise in local chromatin mobility after induction (orange arrow) followed by a return to baseline (green arrow). The increase coincides with elevated active histone marks (H3/ H4 acetylation) and transcription; dynamics subsequently restabilize while oncogene expression and tumor growth persist. Metabolic reprogramming may contribute to this process.

Kim Jaeha, a D4 student of the Genome Diversity Laboratory (Mori Laboratory) at the National Institute of Genetics, received the Best Poster Award at the international symposium 2nd Asian Genetics Consortium Conference (AGCC 2025), held from Friday, November 14 to Sunday, November 16, 2025, at the Numazu City Library.
▶ Award-Winning Poster title:
Behavioral phase transitions in the migratory locust, Locusta migratoria, are related to changes in gut microbiome composition
Maeshima Group / Genome Dynamics Laboratory
Single-nucleosome imaging uncovers biphasic chromatin dynamics in inducible human transformed cells
Aoi Otsuka, Masa A. Shimazoe, Shigeaki Watanabe, Katsuhiko Minami, Sachiko Tamura, Tohru Kiyono, Fumitaka Takeshita, and Kazuhiro Maeshima* (*Corresponding author)
Cell Structure and Function (2025) Advance online publication DOI:10.1247/csf.25147
In eukaryotic cells, genomic DNA folds into nucleosomes to form dynamic domains encompassing euchromatin and heterochromatin. Although many cancer-associated changes in chromatin state and higher-order structure have been reported, how chromatin behavior evolves over time during carcinogenesis has remained unclear.
SOKENDAI students Aoi Otsuka (SOKENDAI Special Researcher) and Masa A. Shimazoe (JSPS DC1), postdoctoral researcher Katsuhiko Minami, technical staff member Sachiko Tamura, and Professor Kazuhiro Maeshima of the Genome Dynamics Laboratory, in collaboration with Visiting Researcher Dr. Tohru Kiyono (also a Visiting Researcher at the Sasaki Institute (Sasaki Foundation)), Project Researcher Dr. Shigeaki Watanabe, and Division Chief Dr. Fumitaka Takeshita at the National Cancer Center, established “EMR” human epithelial cells that inducibly express the oncogenes HPV16 E6/E7, MYC, and KRAS upon doxycycline treatment. Upon induction, EMR cells exhibited cancer-like traits—accelerated proliferation, loss of contact inhibition, soft-agar growth, and tumor formation in nude mice. Live-cell single-nucleosome imaging revealed a biphasic pattern in chromatin dynamics: no change at days 1–3, a transient increase at days 5–7, and a return to baseline by week 4. During this window of increased mobility, histone H3/H4 acetylation and transcription were elevated. Together, these results suggest that oncogene induction causes a transient chromatin “loosening” accompanied by widespread acetylation and transcriptional activation, followed by restabilization of chromatin dynamics even while oncogene expression and tumor growth persist.
This work demonstrates at the single-nucleosome level that the physical behavior of chromatin is reorganized over time during transformation, and that chromatin dynamics can serve as a physical readout of the cancer stage and cellular adaptation.
Funding: JSPS and MEXT KAKENHI (JP23K17398, JP24H00061, JP23KJ0998, JP24KJ1161), JST SPRING JPMJSP2104, and the Takeda Science Foundation.
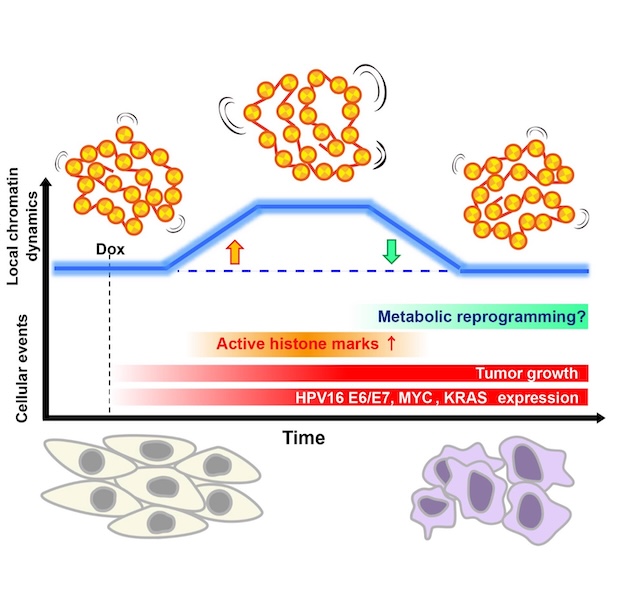
Figure: Model summarizing the biphasic change in local chromatin dynamics during doxycycline-induced transformation of EMR cells. The blue curve denotes a transient rise in local chromatin mobility after induction (orange arrow) followed by a return to baseline (green arrow). The increase coincides with elevated active histone marks (H3/ H4 acetylation) and transcription; dynamics subsequently restabilize while oncogene expression and tumor growth persist. Metabolic reprogramming may contribute to this process.
Press release
Decoupling of visual feature selectivity in the retinocollicular pathway
Ole S. Schwartz, Akihiro Matsumoto, Haruka Yamamoto, and Keisuke Yonehara
Current Biology 2025 DOI:10.1016/j.cub.2025.11.050
![]() Press release (In Japanese only)
Press release (In Japanese only)
The retina is composed of discrete functional cell types that are also characterized by distinct morphology and gene expression. It remains, however, unclear whether similar discrete functional cell types exist in the visual regions downstream of the retina. Here we used two-photon calcium imaging to investigate the response space structure in the retina and in the superficial layers of the mouse superior colliculus (SC), a major retinorecipient area. We found that while retinal ganglion cells showed a clear dependence between responses to luminance and motion, responses to the two stimuli exhibited weaker couplings in collicular neurons. Because of this decoupling, functional clustering based on responses to both luminance and motion had significantly reduced separability compared to clustering based on responses to either. Our work suggests that the SC is not simply a relay station for retinal inputs, but rather generates novel feature selectivity that diversifies cellular responses, perhaps through nonlinear neural processes involving the decoupling and recoupling of retinal ganglion cell’s feature selectivity.
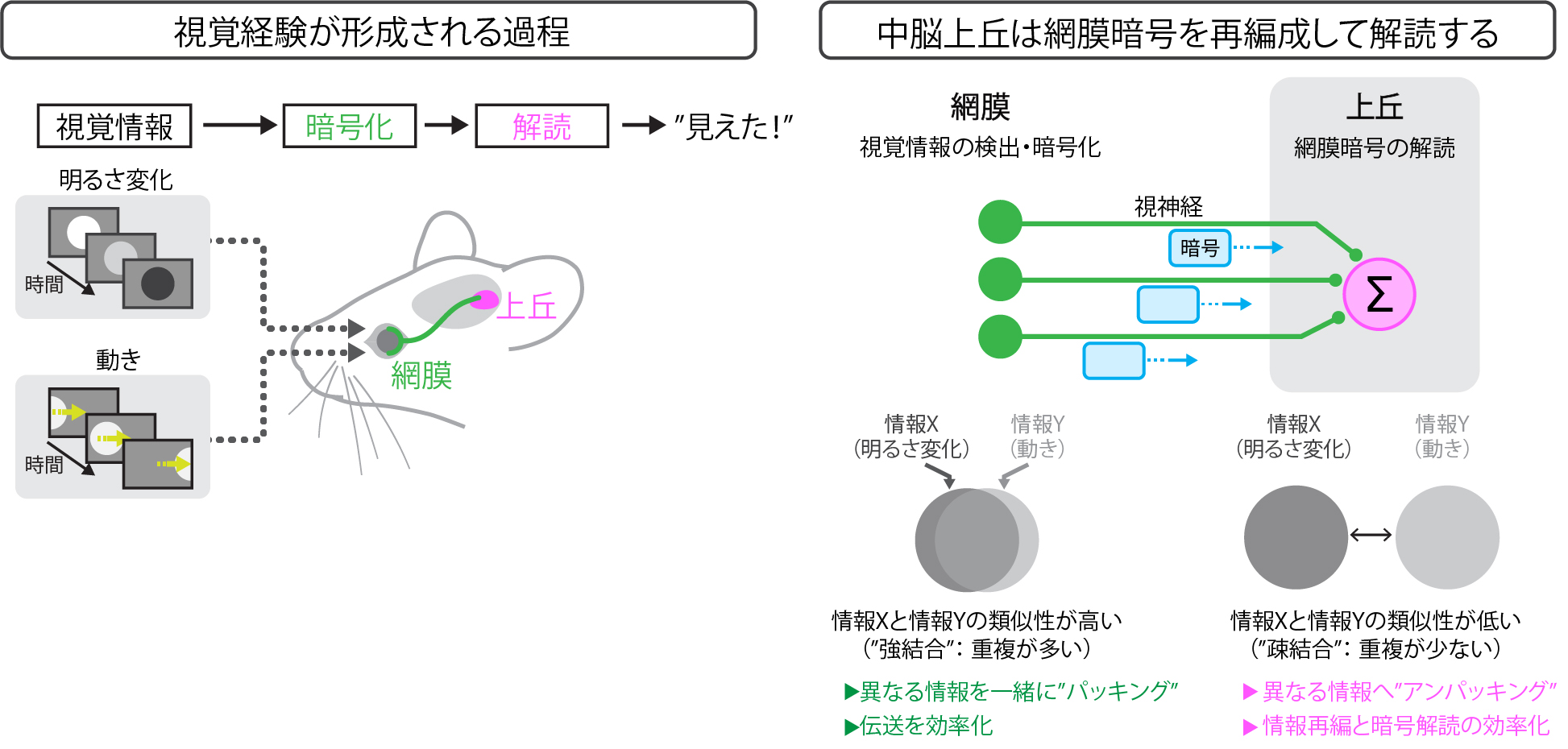
(Left) The visual information processing pathway. Visual information is detected and encoded in the retina and then transmitted to the superior colliculus.
(Right) In the retina, the mutual information between neural activities representing two types of visual information—brightness change (information X) and image motion (information Y)—is high. In the superior colliculus, the redundancy present in the retinal code is decoupled.
Press release
Evolutionary Adaptation of Bacterial proteomes to Translation-Impeding Sequences
Keigo Fujiwara, Naoko Tsuji, Karen Sakiyama, Hironori Niki, and Shinobu Chiba
The EMBO Journal (2025)
![]() Press release (In Japanese only)
Press release (In Japanese only)
In this study, we revealed that diverse bacteria on Earth share amino acid sequences that are difficult for them to synthesize. Because amino acids are essential building blocks of proteins, cells cannot efficiently produce proteins that contain such translation-challenging sequences. Indeed, a comprehensive analysis conducted across the bacterial domain showed that these translation-challenging sequences are rarely found within bacterial proteins, suggesting that bacteria have evolutionarily avoided using them.
In contrast, we also discovered that these sequences frequently appear near the ends (carboxyl termini) of relatively small proteins. Bioinformatic analyses further indicated that such proteins likely play important and diverse roles in helping cells adapt to fluctuating environmental conditions.
Together, this work demonstrates that translation-challenging sequences, although generally disadvantageous and evolutionarily excluded from most proteins, can be repurposed by bacteria as functional elements. By leveraging the “difficulty” of translating these sequences, various bacteria appear to have evolved unique strategies to better cope with environmental changes.
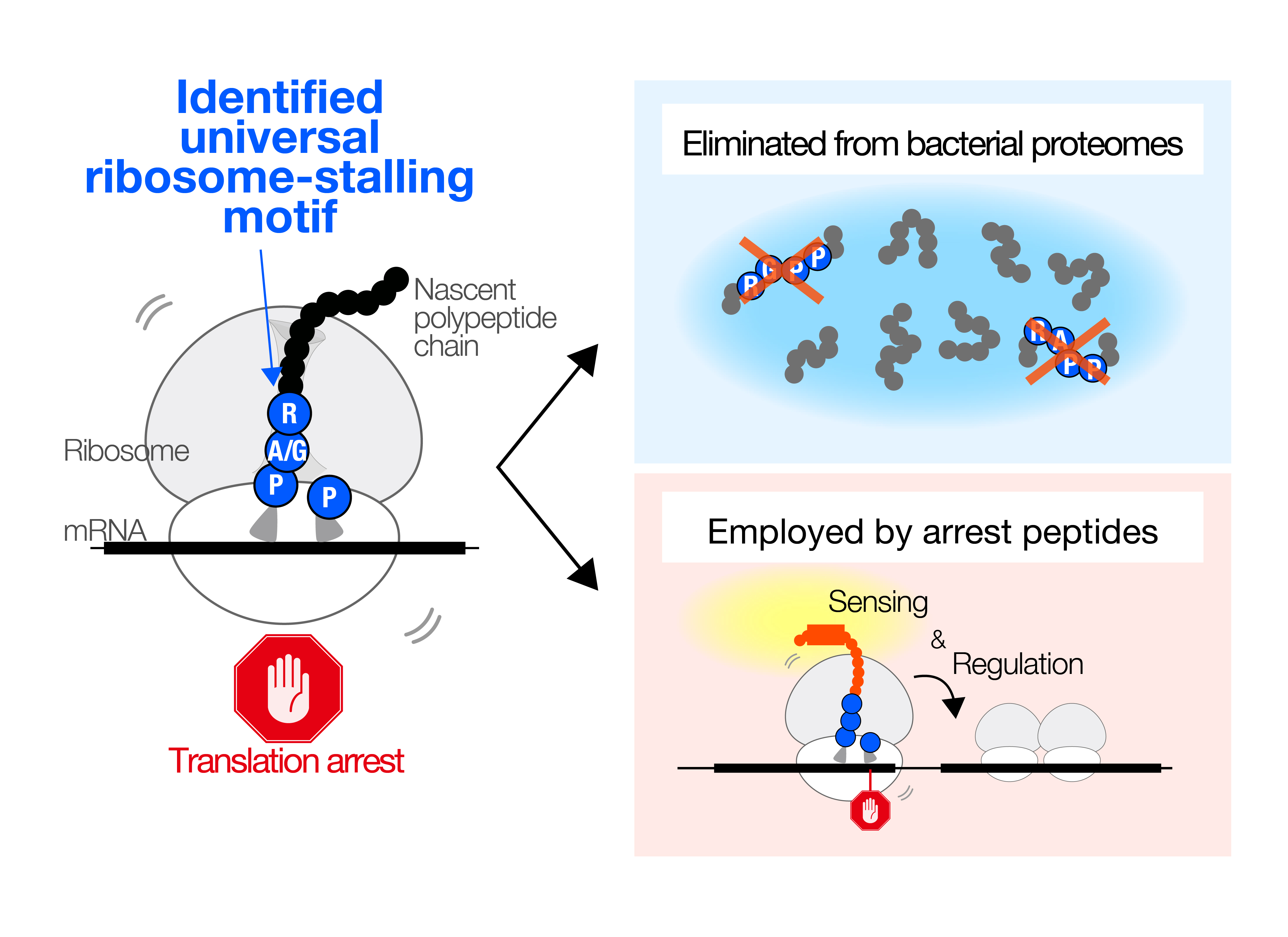
Figure:We identified patterns of “translation arrest-causing amino acid sequences” that occur across many bacteria (left). Although such sequence patterns are generally eliminated during evolution (upper right), some bacteria have instead evolved unique mechanisms that leverage this inherent synthesis difficulty to support important cellular functions (lower right)
Press release
Niki Group / Microbial Physiology Laboratory
Evolutionary Adaptation of Bacterial proteomes to Translation-Impeding Sequences
Keigo Fujiwara, Naoko Tsuji, Karen Sakiyama, Hironori Niki, and Shinobu Chiba
The EMBO Journal (2025)
In this study, we revealed that diverse bacteria on Earth share amino acid sequences that are difficult for them to synthesize. Because amino acids are essential building blocks of proteins, cells cannot efficiently produce proteins that contain such translation-challenging sequences. Indeed, a comprehensive analysis conducted across the bacterial domain showed that these translation-challenging sequences are rarely found within bacterial proteins, suggesting that bacteria have evolutionarily avoided using them.
In contrast, we also discovered that these sequences frequently appear near the ends (carboxyl termini) of relatively small proteins. Bioinformatic analyses further indicated that such proteins likely play important and diverse roles in helping cells adapt to fluctuating environmental conditions.
Together, this work demonstrates that translation-challenging sequences, although generally disadvantageous and evolutionarily excluded from most proteins, can be repurposed by bacteria as functional elements. By leveraging the “difficulty” of translating these sequences, various bacteria appear to have evolved unique strategies to better cope with environmental changes.
Figure:We identified patterns of “translation arrest-causing amino acid sequences” that occur across many bacteria (left). Although such sequence patterns are generally eliminated during evolution (upper right), some bacteria have instead evolved unique mechanisms that leverage this inherent synthesis difficulty to support important cellular functions (lower right)
Koide Group / Mouse Genomics Resource Laboratory
Chromosome-scale genomes of two wild flowering cherries (Cerasus itosakura and C. jamasakura) provide insights into structural evolution in Cerasus
Kazumichi Fujiwara, Atsushi Toyoda, Toshio Katsuki, Yutaka Sato, Bhim B Biswa, Takushi Kishida, Momi Tsuruta, Yasukazu Nakamura, Takako Mochizuki, Noriko Kimura, Shoko Kawamoto, Tazro Ohta, Ken-Ichi Nonomura, Hironori Niki, Hiroyuki Yano, Kinji Umehara, Chikahiko Suzuki, Tsuyoshi Koide
DNA Research (2025) DOI:10.1093/dnares/dsaf031
Japan’s iconic cherry trees hold deep cultural significance, yet chromosome-scale genomic resources for their wild relatives have been lacking. The Sakura 100 Genome Consortium, led by the National Institute of Genetics and the Forestry and Forest Products Research Institute, has now produced the first high-quality, chromosome-scale genomes of Cerasus itosakura (Edohigan) and C. jamasakura (Yamazakura).
The genomes show exceptional completeness and reveal both broad conservation and key species differences. Notably, a 1.84-Mb inversion on chromosome 8 was identified in C. itosakura, providing new insight into lineage-specific chromosomal evolution. Differences in rRNA gene cluster organization further highlight genomic diversity within Cerasus.
Using these new references, the team reconstructed the haplotypes of the ornamental cultivar ‘Somei-yoshino’, confirming its hybrid origin from C. itosakura and C. speciosa.
This work advances the understanding of cherry evolution and offers a crucial foundation for breeding, taxonomy, and conservation.

A–B : Flowers and trees of C. itosakura. C-D:Flowers and trees of C. jamasakura. E: Chromosome-level genome structures of C. jamasakura and C. itosakura. F: A large inversion identified on chromosome 8 of C. itosakura. This inversion is not observed in C. speciosa, C. jamasakura, or C. campanulata.
This work was supported by grants from ROIS (Research Organization of Information and Systems).
Press release
Regulated TRESLIN-MTBP loading governs initiation zones and replication timing in human DNA replication
Xiaoxuan Zhu, Atabek Bektash, Yuki Hatoyama, Sachiko Muramatsu, Shin-Ya Isobe, Chikashi Obuse, Atsushi Toyoda, Yasukazu Daigaku, Chun-Long Chen and Masato T. Kanemaki
Nature Communications 2025 DOI:10.1038/s41467-025-66278-7
![]() Press release (In Japanese only)
Press release (In Japanese only)
When cells proliferate, genomic DNA is precisely duplicated once per cell cycle. Abnormalities in this DNA replication process can cause alterations in genomic DNA, contributing to cellular ageing, cancer, and hereditary diseases. Therefore, understanding how cells replicate their DNA is crucial for elucidating fundamental biological processes, diseases, and even evolution.
Traditionally, DNA replication has been studied in microorganisms such as E. coli and yeast. In these organisms, the locations where DNA replication begins (replication origins) are determined by specific DNA sequences. However, in most eukaryotic cells, including human cells, the DNA sequence itself does not dictate where replication starts. For decades, it remained a mystery how and where replication is initiated within the human genome.
To address this, Professor Masato Kanemaki and his team including the first author, Dr Xiaoxuan Zhu, at National Institute of Genetics developed a new high-precision method, LD-OK-seq (Ligase Depletion-Okazaki sequencing), to detect replication initiation regions in the human genome. By further analysing the proteins bound to these regions, they uncovered the fundamental principle by which human cells determine replication initiation sites.
Their findings revealed that, except for actively transcribed gene regions, human cells possess the ability to initiate DNA replication from almost anywhere in the genome. This capability arises from the widespread binding of an enzyme called the MCM helicase, which is essential for DNA replication. Moreover, they discovered that during the early S phase, replication frequently begins in intergenic regions (areas between transcribed genes), and that these sites are determined by the binding of TRESLIN-MTBP, a protein complex that activates the MCM helicase. They also identified an antagonistic regulatory system that modulates the binding of TRESLIN-MTBP to MCM.
These discoveries answer the fundamental question of how human cells initiate genome replication, providing new insights into diseases caused by replication abnormalities—such as genomic instability disorders, cancer, aging, and hereditary diseases—as well as into genome evolution. In the long term, this work may also lay the foundation for technologies that enable artificial control of DNA replication.
This study was conducted through an international collaboration between the research group of Professor Masato Kanemaki and Project Professor Atsushi Toyoda at the National Institute of Genetics, Professor Chikashi Obuse at The University of Osaka, Dr. Yasukazu Daigaku at the Cancer Institute of JFCR, and Professor Chun-Long Chen at Curie Institute in France. The research was supported by JSPS KAKENHI grants (JP23H02463, JP21H04719, JP23H04925, JP25H00979), Platform for Advanced Genome Science (JP22H04925), JST FOREST (JPMJFR204X), JST CREST (JPMJCR21E6), and AMED ASPIRE (JP25jf0126015).
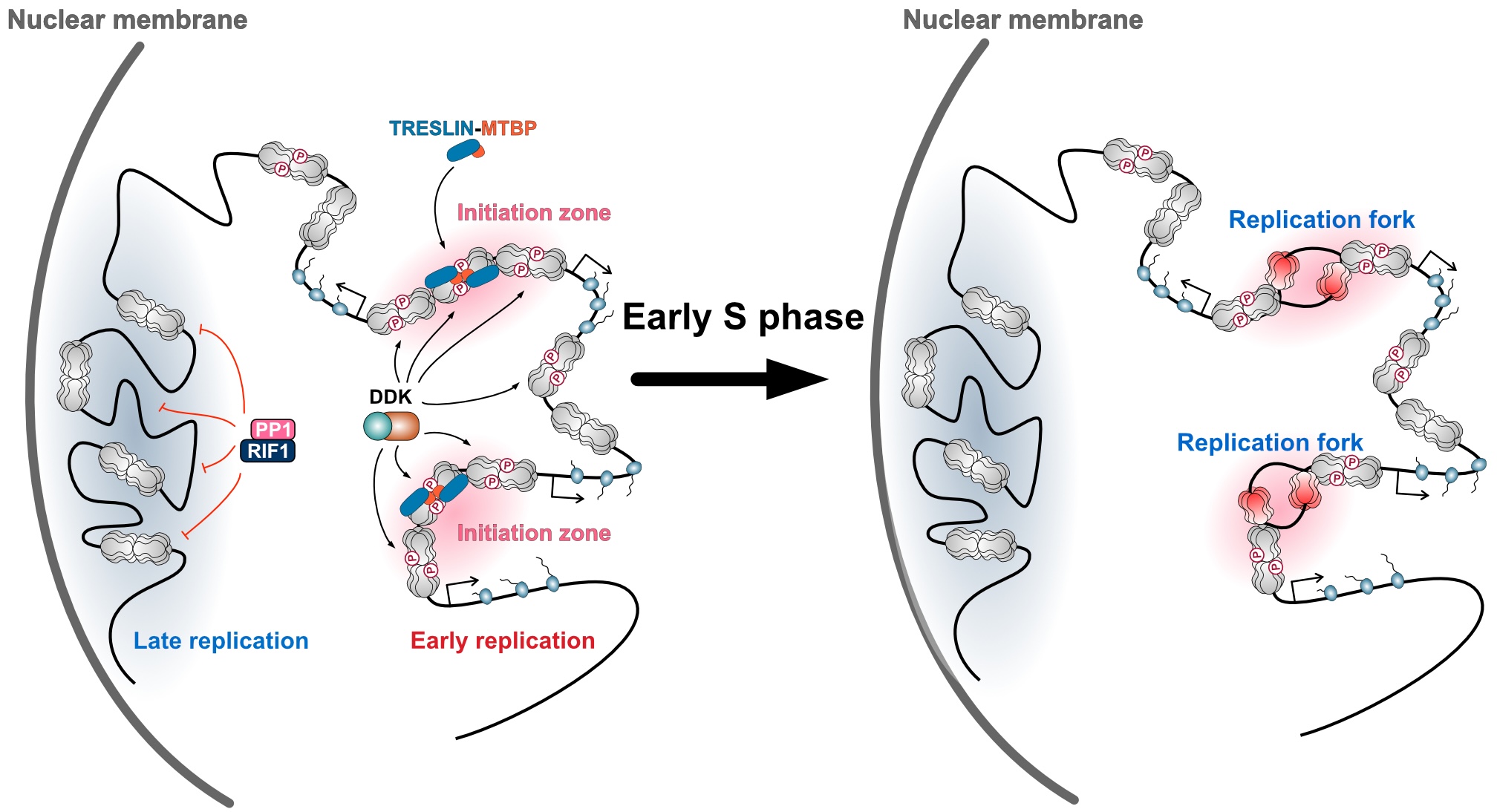
Figure: The MCM helicase is broadly bound across genomic DNA in regions outside actively transcribed genes, and its phosphorylation is antagonistically regulated by the kinase DDK and the phosphatase RIF1–PP1. The sites of replication initiation are determined when the TRESLIN–MTBP complex is recruited to the phosphorylated MCM helicase.










