



Mammalian Development Laboratory / Saga Group
Sfrp5 identifies murine cardiac progenitors for all myocardial structures except for the right ventricle
Fujii Masayuki, Akane Sakaguchi, Ryo Kamata, Masataka Nagao, Shilvia M. Evans, Masao Yoshizumi, Akihiko Shimono, Yumiko Saga, and Hiroki Kokubo
Nature Communications. DOI:10.1038/NCOMMS1466
Upon acquirement of pulmonary circulation, the ancestral heart may have been remodelled coincidently with, or accompanied by, the production and rearrangement of progenitor cells. However, the progenitor populations that give rise to the left ventricle (LV) and sinus venosus (SV) are still ambiguous. Here we show that the expression of Secreted frizzled-related protein Sfrp5 in the mouse identifies common progenitors for the outflow tract (OFT), LV, atrium and SV but not the right ventricle (RV). Sfrp5 expression begins at the lateral sides of the cardiac crescent, excluding early differentiating regions, and continues in the venous pole, which gives rise to the SV. Lineage-tracing analysis revealed that descendants of Sfrp5-expressing cells at E7.5 contribute not only to the SV but also to the LV, atria and OFT and are found also in the dorsal splanchnic mesoderm accompanied by the expression of the secondary heart field marker, Islet1. These findings provide insight into the arrangement of cardiac progenitors for systemic circulation. This study was conducted as a collaboration research with Dr. Kokubo who used to be an assistant professor in Mammalian Development Lab and currently a lecturer of Hiroshima University.
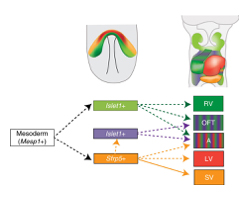
Upper panels show the expression area ofSfrp5 (orange) andIslet1 (light green) at E7.5 (left) and the distribution of descendant cells at E9.5 (right).
The lower panel shows the lineage contribution of the heart. Mesp1-positive mesodermal cells are common cardiac precursors.TransientIslet1-expressing cells that do not expressSfrp5 contribute to the RV, OFT and atrium (green). Sfrp5-positive cells contribute to the LV, atrium and OFT (orange) the SV
On March 14, 2017 two students who graduated in March 2016 received Morishima Awards from the Department of Genetics, SOKENDAI. The Morishima Award was established using a donation from the Morishima family honoring the late professor emeritus of NIG, Dr. Hiroko MORISHIMA. It awards students who receive a doctoral degree from the Department of Genetics to acknowledge their outstanding performances during PhD studies.
・Yuki TSUCHIYA (Division of Centrosome Biology, thesis advisor Daiju KITAGAWA)
thesis title : The role of Cep295 in centrosome biogenesis of human cells
・Kazunori YAMAMTO (Cell Architecture Laboratory, thesis advisor: Akatsuki KIMURA)
thesis title : A study on inter-cellular forces for blastomere configurations in developing embryos

Press release
Endoplasmic Reticulum-Mediated Microtubule Alignment Governs Cytoplasmic Streaming
Kenji Kimura, Alexandre Mamane, Tohru Sasaki, Kohta Sato, Jun Takagi, Ritsuya Niwayama, Lars Hufnagel, Yuta Shimamoto, Jean-François Joanny, Seiichi Uchida, and Akatsuki Kimura
Nature Cell Biology (2017) DOI:10.1038/ncb3490
Pressrelease (In Japanese only)
A research group lead by Drs. Kenji Kimura and Akatsuki Kimura at NIG revealed the self-organization mechanism of cytoplasmic streaming in Caenorhabditis elegans zygotes. Cytoplasmic streaming refers to a collective movement of cytoplasm observed in many cell types. The mechanism of meiotic cytoplasmic streaming (MeiCS) in C. elegans zygotes was puzzling as the direction of the flow is not predefined by cell polarity and occasionally reverses. The research group demonstrated that the endoplasmic reticulum (ER) network structure is required for the collective flow (Fig. 1). Using a combination of RNAi, microscopy, and image processing of C. elegans zygotes, the group devised a theoretical model, which reproduced and predicted the emergence and reversal of the flow. They proposed a positive feedback mechanism, where a local flow generated along a microtubule is transmitted to neighboring regions through the ER (Fig. 2). This, in turn, aligns microtubules over a broader area to self-organize the collective flow. The proposed model could be applicable to various cytoplasmic streaming phenomena in the absence of predefined polarity. The increased mobility of cortical granules by MeiCS correlated with the efficient exocytosis of the granules to protect the zygotes from osmotic and mechanical stresses.
The research was conducted as collaboration between NIG (Cell Architecture lab and Quantitative Mechanobiology lab), Kyushu University, Institut Curie (France) and EMBL (Germany).
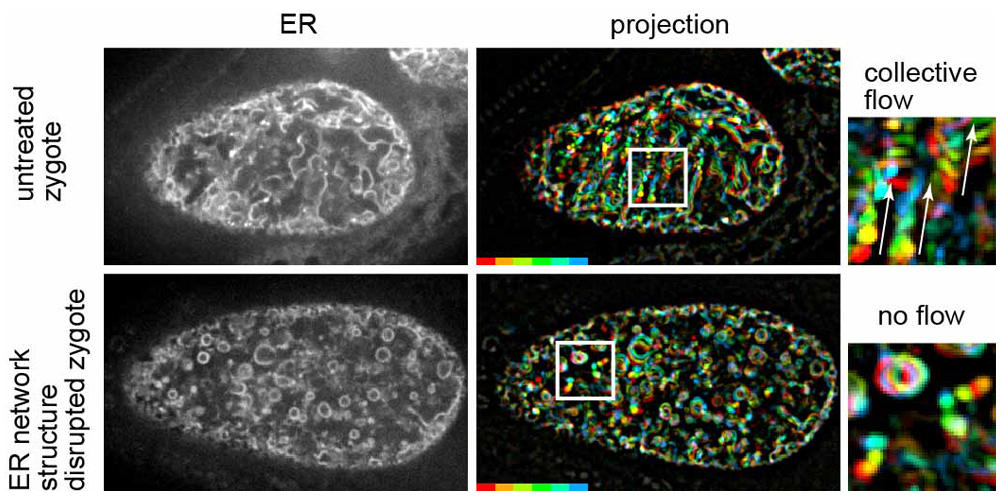
Fig. 1
Left panels show fluorescence confocal images of the ER during MeiCS in the C. elegans zygote (upper: untreated zygote, bottom: RNAi-treated zygote in which the network structure of the ER was fragmented). Middle panels show projections of sequential images of the ER. Fluorescence signals are colored to indicate their trajectories. Fragmentation of the ER network inhibited the flow. Boxed region is magnified on the right. White arrows indicate the flow direction.
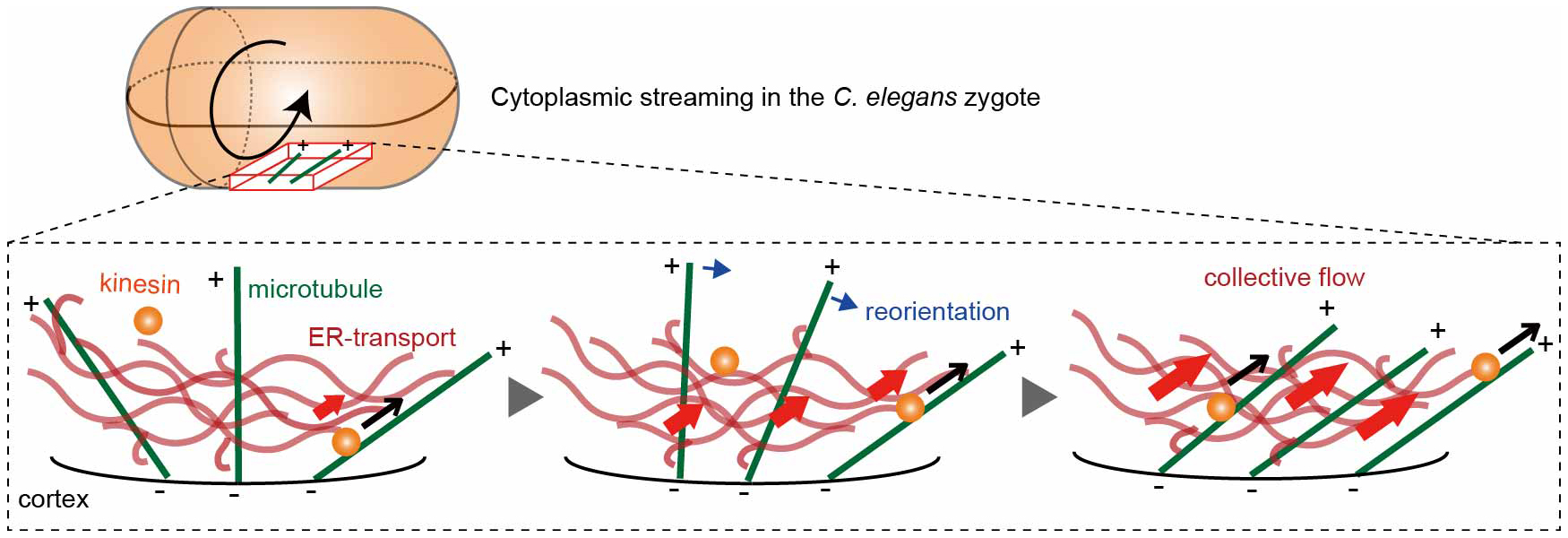
Fig. 2
ER-mediated positive feedback model. When kinesin transports the ER network along a cortical microtubule (black arrows), neighboring microtubules are biased to reorient (blue arrows) toward the ER motion, enhancing collective flow (red arrows). (−) and (+) signs indicate microtubule polarity.
Multicellular Organization Laboratory / Sawa Group
PIGN prevents protein aggregation in the endoplasmic reticulum independently of its function in the GPI synthesis
Shinji Ihara, Sohei Nakayama, Yoshiko Murakami, Emiko Suzuki, Masayo Asakawa, Taroh Kinoshita and Hitoshi Sawa
J Cell Sci 2017 130: 602-613; DOI:10.1242/jcs.196717
1. Protein aggregation is a common feature of many neurodegenerative diseases and often results from defective folding processes. We found that PIGN functions in protein quality control to prevent protein aggregation within the ER in C. elegans and in mammalian cells. Although PIGN was originally identified as an enzyme involved in glycosylphosphatidylinositol (GPI)-anchor biosynthesis, we show that the function of PIGN in protein quality control is independent of its enzymatic activities. In human, several mutations in PIGN have been shown to cause multiple congenital anomalies-hypotonia-seizures syndrome 1 (MCAHS1) that shows dysmorphic facial features and Fryns syndrome that cause lethality in the neonatal period. C. elegans with pign mutations created by CRISPR/Cas9 that correspond to MCAHS1 patients also cause protein aggregation in the ER, implying that the dysfunction of the PIGN non-canonical function might affect symptoms of MCAHS1 and potentially those of other diseases.
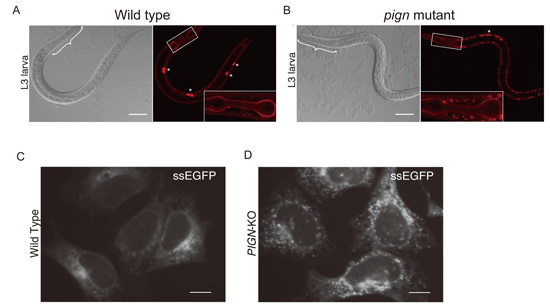
Accumulation of secretory proteins in C. elegans and mammalian cells with PIGN mutations.
(A) The localization of collagen-IV::mCherry (right) and differential interference contrast (DIC) images (left) of wild-type (A) and PIGN mutant (B) C. elegans.
The localization of secreted soluble EGFP (ssEGFP) in wild-type (C) and PIGN-knockout (D) HEK293 cells.
New assistant professors join NIG as of March 1, 2017.
![]()
Evolution of Shh endoderm enhancers during morphological transition from ventral lungs to dorsal gas bladder
Tomoko Sagai, Takanori Amano, Akiteru Maeno, Tetsuaki Kimura, Masatoshi Nakamoto, Yusuke Takehana, Kiyoshi Naruse, Norihiro Okada, Hiroshi Kiyonari, Toshihiko Shiroishi
Nature communications 8, Article number: 14300 (2017) DOI:10.1038/ncomms14300
Pressrelease (In Japanese only)
The lung and the gas bladder are most likely homologous organs, and now the gas bladder is regarded as an evolutionary modification of the lung. Transition of the lungs to the non-respiratory gas bladder for buoyancy control was crucial for the aquatic life. Because it is inferred that molecular changes in cis-regulatory element (CRE) of pleiotropic developmental genes have more often contributed to evolution of vertebrate body plan than changes in coding sequences, in this study we intended to elucidate evolution of enhancers, which was associated with morphological transition from the lungs to the gas bladder.
Shh signaling plays a crucial role for endoderm development. A Shh endoderm enhancer, MACS1, is well conserved across terrestrial animals with lungs. First, we showed that elimination of mouse MACS1 causes severe defects in laryngeal development, leading to postnatal lethality. It indicated that MACS1-directed Shh signalling is indispensable for respiratory organogenesis. Extensive phylogenetic analyses revealed that MACS1 emerged prior to the divergence of cartilaginous and bony fishes, and even euteleost fishes have a MACS1 orthologue. Meanwhile, ray-finned fishes evolved a novel conserved non-coding sequence in the neighbouring region. Transgenic assays showed that MACS1 drives reporter expression ventrally in laryngeal epithelium. This activity has been lost in the euteleost lineage, and instead, the conserved non-coding sequence of euteleosts acquired an enhancer activity to elicit dorsal epithelial expression in the posterior pharynx and oesophagus. These results implicate that evolution of these two enhancers is relevant to the morphological transition from ventral lungs to dorsal gas bladder.
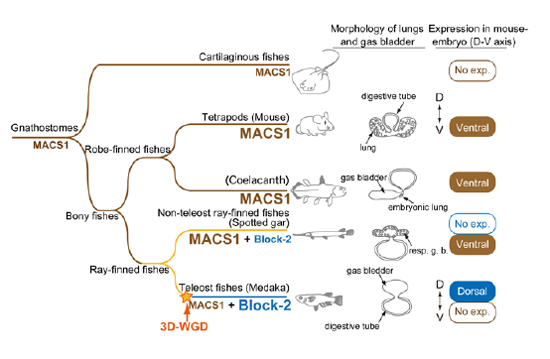
Evolution of two Shh enhancers in vertebrates
Ancestral and inactive form of MACS1 appeared in the common ancestor prior to separation of lobe-finned and ray-finned fish lineages. In the tetrapods and non-teleost ray-finned fish, the inactive MACS1 evolved to the active form, which drives the Shh expression in the ventral epithelium of the prospective laryngeal structure. It is considered that the MACS1 sequence largely diverged and lost the intrinsic activity in the teleost fishes. In the meantime, ancestral form of Block-2 appeared among the non-teleost ray-finned fishes. Teleost fishes evolved the primitive Block-2 into the active form that drives the Shh expression in the dorsal epithelium of the oesophagus. resp. g. b., respiratory gas bladder; No exp., No expression.

On Thursday, 26th January 2017, Dr. Junichi Tomizawa, the 6th Director-General of National Institute of Genetics passed away at the age of 92.
In honor of his memory and great contributions to science, we would like to express our sincere condolences.
Division of Agricultural Genetics / Kakutani Group
Gene-body chromatin modification dynamics mediate epigenome differentiation in Arabidopsis
Soichi Inagaki, Mayumi Takahashi, Aoi Hosaka, Tasuku Ito, Atsushi Toyoda, Asao Fujiyama, Yoshiaki Tarutani, Tetsuji Kakutani
The EMBO Journal. Published online 18.01.2017 DOI:10.15252/embj.201694983
Heterochromatin is marked by methylation of lysine 9 on histone H3 (H3K9me). A puzzling feature of H3K9me is that this modification localizes not only in promoters but also in internal regions (bodies) of silent transcription units. Despite its prevalence, the biological significance of gene-body H3K9me remains enigmatic. Here we show that H3K9me-associated removal of H3K4 monomethylation (H3K4me1) in gene bodies mediates transcriptional silencing. Mutations in an Arabidopsis H3K9 demethylase gene IBM1 induce ectopic H3K9me2 accumulation in gene bodies, with accompanying severe developmental defects. Through suppressor screening of the ibm1-induced developmental defects, we identified the LDL2 gene, which encodes a homolog of conserved H3K4 demethylases. The ldl2 mutation suppressed the developmental defects, without suppressing the ibm1-induced ectopic H3K9me2. The ectopic H3K9me2 mark directed removal of gene-body H3K4me1 and caused transcriptional repression in an LDL2-dependent manner. Furthermore, mutations of H3K9 methylases increased the level of H3K4me1 in the gene bodies of various transposable elements, and this H3K4me1 increase is a prerequisite for their transcriptional derepression. Our results uncover an unexpected role of gene-body H3K9me2/H3K4me1 dynamics as a mediator of heterochromatin silencing and epigenome differentiation.
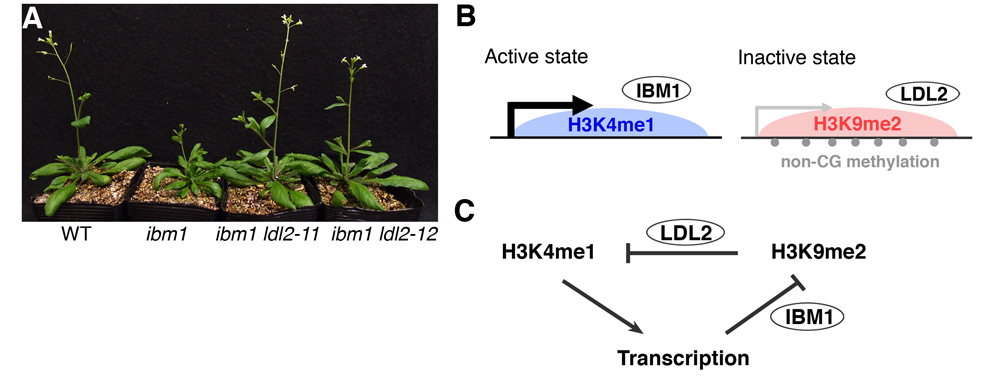
(A) ibm1 ldl2 double mutants show suppression of developmental abnormalities in the ibm1 mutant. (B) Active and inactive chromatin states are determined by two histone demethylases, IBM1 and LDL2. (C) A model for differentiation of bi-stable epigenetic states with feedback regulation. Transcription induces H3K9 demethylation by IBM1 and maintains active chromatin state. Conversely, accumulation of H3K9me2 induces demethylation of H3K4me1 and represses transcription.
DBCLS
Comparative Genomics Laboratory
Advanced Genomics Center
Identification of functional enolase genes of the silkworm Bombyx mori from public databases with a combination of dry and wet bench processes
Akira Kikuchi, Takeru Nakazato, Katsuhiko Ito, Yosui Nojima, Takeshi Yokoyama, Kikuo Iwabuchi, Hidemasa Bono, Atsushi Toyoda, Asao Fujiyama, Ryoichi Sato and Hiroko Tabunoki
BMC Genomics, 18:83, 2017 DOI:10.1186/s12864-016-3455-y
Researchers can now easily obtain genomic information from online databases. However, many incorrectly annotated genes are included in these databases, which can prevent the correct interpretation of subsequent functional analyses. To address this problem, we used a combination of dry and wet bench processes to identify functional enolases in the silkworm Bombyx mori.
To identify enolase sequences in B. mori, we performed a HMM search of public databases. We found five enolase sequences, which we then characterized using RNA-seq analysis, cDNA cloning, and RT-PCR. Finally, we determined that three enolase genes in B. mori were functional. Our strategy could be helpful for the detection of minor genes and functional genes in non-model organisms from public databases.
Prof. Atsushi Toyoda and Prof. Asao Fujiyama (Center for Information Biology) contributed to sequencing testis transcriptome of B. mori. Dr. Takeru Nakazato and Dr. Hidemasa Bono (Database Center for Life Science) contributed to this work in dry analysis.
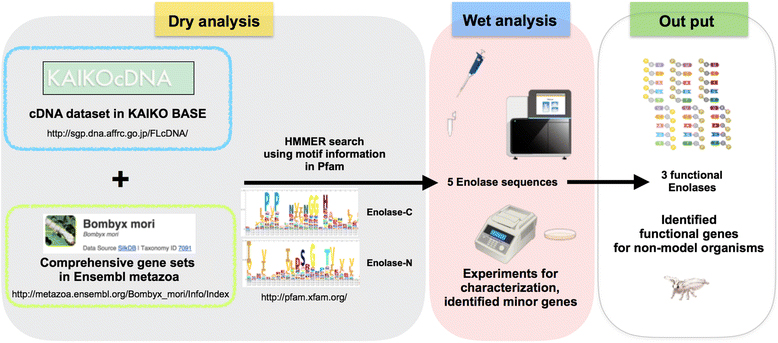
Combining dry and wet bench processes to identify functional enolases in the silkworm B. mori. To identify enolase sequences in B. mori, we performed a HMM search of public databases. We found five enolase sequences, which we then characterized using RNA-seq analysis, cDNA cloning, and RT-PCR. Finally, we determined that three enolase genes in B. mori were functional.










