



Division of Symbiosis and Cell Evolution / Miyagishima Group
Chloroplast division checkpoint in eukaryotic algae.
Nobuko Sumiya, Takayuki Fujiwara, Atsuko Era, and Shin-ya Miyagishima
Proc. Natl. Acad. Sci. USA. (2016). Published online, DOI:10.1073/pnas.1612872113
Chloroplasts arose from a cyanobacterial endosymbiont, which introduced photosynthesis into eukaryotes. It is widely believed that synchronization of division in the eukaryotic host cell and in the endosymbiont was critical for the host cell to maintain the endosymbiont/chloroplast permanently. However, it is unclear how the division of the endosymbiont (the chloroplast) and host cell became synchronized. Using the unicellular red alga Cyanidioschyzon merolae, we show that the host cell enters into the metaphase only when chloroplast division has commenced. A similar phenomenon also was observed in the glaucophyte alga Cyanophora paradoxa. It thus seems likely that the acquisition of the cell-cycle checkpoint of chloroplast division played an important role in the establishment of the chloroplast in ancient algae.
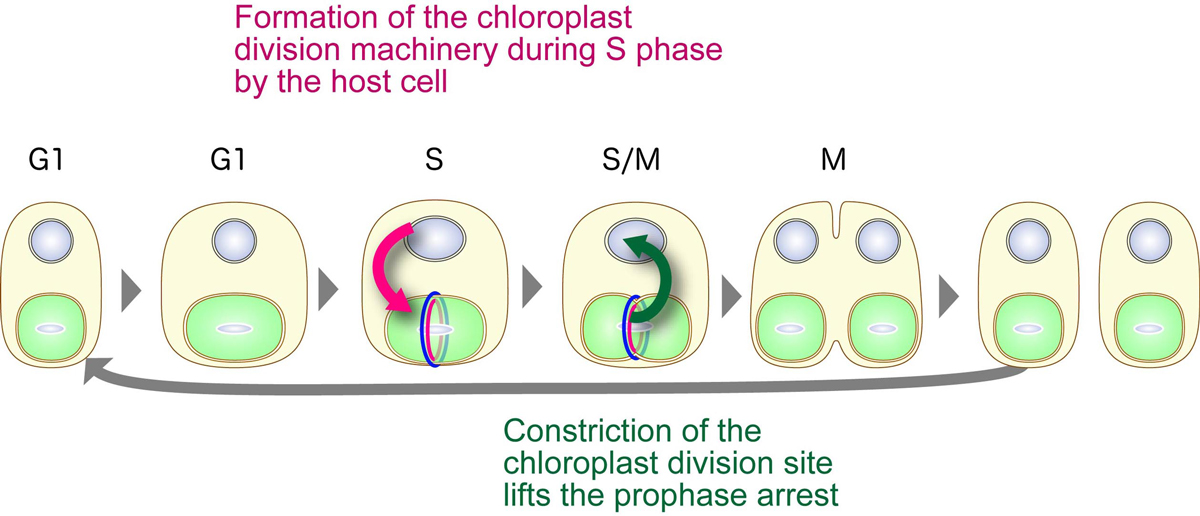
The interactive synchronization of division in the eukaryotic host cell and the chloroplast in algae. The host cell restricts the onset of chloroplast division to the S phase by S-phase–specific expression of nucleus-encoded chloroplast-division proteins. The formation of the competent chloroplast-division machinery and constriction of the chloroplast division site lift the prophase arrest so that the host cell enters into the metaphase only when chloroplast division progresses.
![]()
Dynamic nucleosome movement provides structural information of topological chromatin domains in living human cells
Soya Shinkai, Tadasu Nozaki, Kazuhiro Maeshima, Yuichi Togashi
PLOS Computational Biology. 12(10), e1005136 DOI:10.1371/journal.pcbi.1005136
Pressrelease (In Japanese only)
The mammalian genome is organized into submegabase-sized chromatin domains (CDs) including topologically associating domains, which have been identified using chromosome conformation capture-based methods. Single-nucleosome imaging in living mammalian cells has revealed subdiffusively dynamic nucleosome movement (left in Fig.). It is unclear how single nucleosomes within CDs fluctuate and how the CD structure reflects the nucleosome movement. Here, we present a polymer model wherein CDs are characterized by fractal dimensions and the nucleosome fibers fluctuate in a viscoelastic medium with memory. We analytically show that the mean-squared displacement (MSD) of nucleosome fluctuations within CDs is subdiffusive. The diffusion coefficient and the subdiffusive exponent depend on the structural information of CDs. This analytical result enabled us to extract information from the single-nucleosome imaging data for HeLa cells (right in Fig.). Our observation that the MSD is lower at the nuclear periphery region than the interior region indicates that CDs in the heterochromatin-rich nuclear periphery region are more compact than those in the euchromatin-rich interior region with respect to the fractal dimensions as well as the size. Finally, we evaluated that the average size of CDs is in the range of 100–500 nm and that the relaxation time of nucleosome movement within CDs is a few seconds. Our results provide physical and dynamic insights into the genome architecture in living cells.
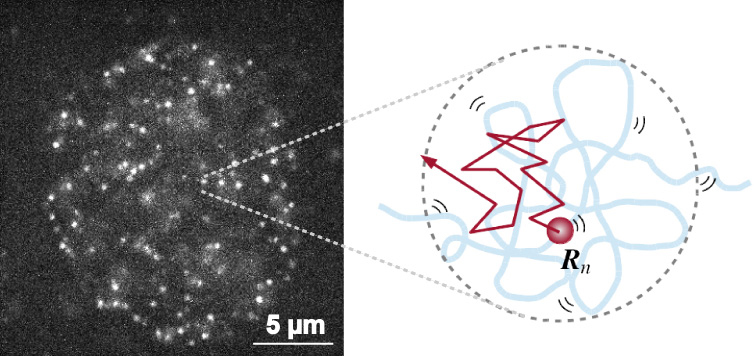
Single-nucleosome image in a living human nucleus (left). Nucleosome dynamically fluctuates within the chromatin domain and shows subdiffusion (right). By applying the mathematical model to the nucleosome movement, we can obtain the information on chromatin structure.
The Research Organization of Information and Systems announced the ROIS Education and Research Council approved the reappointment of Dr. Isao Katsura who will expire his term of office on 30th of November, 2016 to the office of Director General, National Institute of Genetics.
His reappointment is effective from 1st of December, 2016 for a two year term.
![]()
Sexual Fate Change of XX Germ Cells Caused by the Deletion of SMAD4 and STRA8 Independent of Somatic Sex Reprogramming
Quan Wu, Kurumi Fukuda, Yuzuru Kato, Zhi Zhou, Chu-Xia Deng, Yumiko Saga
PLOS Biology September 8, 2016 DOI:10.1371/journal.pbio.1002553
Pressrelease (In Japanese only)
Mammalian sex depends on a male-specific gene, sex-determining region Y (SRY), which is located on the Y chromosome. Individuals lacking this gene will develop as female. Accordingly, germ cell fate also changes from male to female in the absence of SRY. Therefore, it is thought that somatic cells regulate germ cells to become sperm or oocytes. However, it is largely unknown what factor is responsible for sexual fate determination in germ cells. In fetal ovaries, retinoic acid (RA) initiates STRA8 expression in germ cells and induces meiosis. Female germ cells without STRA8 fail to enter meiosis but still progress to oogenesis and form oocyte-like cells, indicating that RA is not the regulator of oogenesis. Here, we found that female germ cells lacking both SMAD4 and STRA8 (but not a single knockout) develop as male gonocyte-like cells in ovaries, indicating that these two factors work as female germ cell determinants. To our surprise, the sexual fate switch observed in the double knockout ovary is not accompanied by gene expression changes in somatic cells, revealing the unexpected finding that somatic factors controlled by SRY are dispensable for the upregulation of male-specific genes in germ cells. This research is partly supported by Grant-in-Aid for Scientific Research on Innovative Areas ”Epigenome dynamics and regulation in germ cells” to YS.
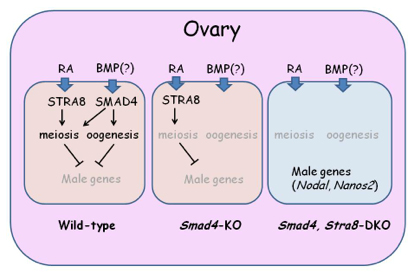
In the ovary, two pathways, RA-Stra8 and SMAD4 pathways play crucial roles. In the absence of SMAD4, germ cells fail to produce any oocytes but do not show any male character. Loss of SMAD4 and STRA8 leads to the upregulation of NANOS2 as well as other male-specific genes.
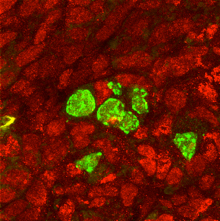
Smad4/Stra8-dKO germ cells express a male factor PLZF (green) in the ovary filled with female somatic cells (marked by Foxl2)。
Experimental Farm / Nonomura Group
Histone H3 modifications are widely reprogrammed during male meiosis I in rice dependently on MEL1 Argonaute protein
Hua Liu, Ken-Ichi Nonomura
Journal of Cell Science, Published online, 12 August, 2016 DOI:10.1242/jcs.184937
Meiosis is a special type of cell division to halve the chromosome number, achieved by two continuous division not intervened by DNA replication. It is an indispensable mechanism to generate genetic diversity via homologous chromosome pairing and meiotic recombination, in addition to stable transmission of genetic information to the next generation.
We focused on the relation of meiosis and histone modifications (glossary), which are important for control of chromosome structure and gene expression. Generally in plants, dimethylation at the position-9 lysine of histone H3 (H3K9me2) is thought to repress gene expressions and promote the compaction of chromatin structure. In contrast, acethylation at the same position (H3K9ac) activates gene expressions. We found that H3 modifications after the meiotic entry were totally altered from the premeiotic H3 status (Fig. 1A). “Large-scale meiotic chromosome reprogramming (LMR)” named in this paper is thought to be one of the mechanisms promoting meiosis in plants.
Interestingly, LMR was completely disrupted in the mutant of MEL1 (Fig. 1B), that is an Argonaute protein (glossary) specifically expressed in rice germ cells. These results suggest possibilities that MEL1 promotes meiosis via control of LMR, and that the RNA silencing mechanism is important for plant meiosis.
This work was supported by JSPS KAKENHI (25252004), and by NIG postdoc fellowship.
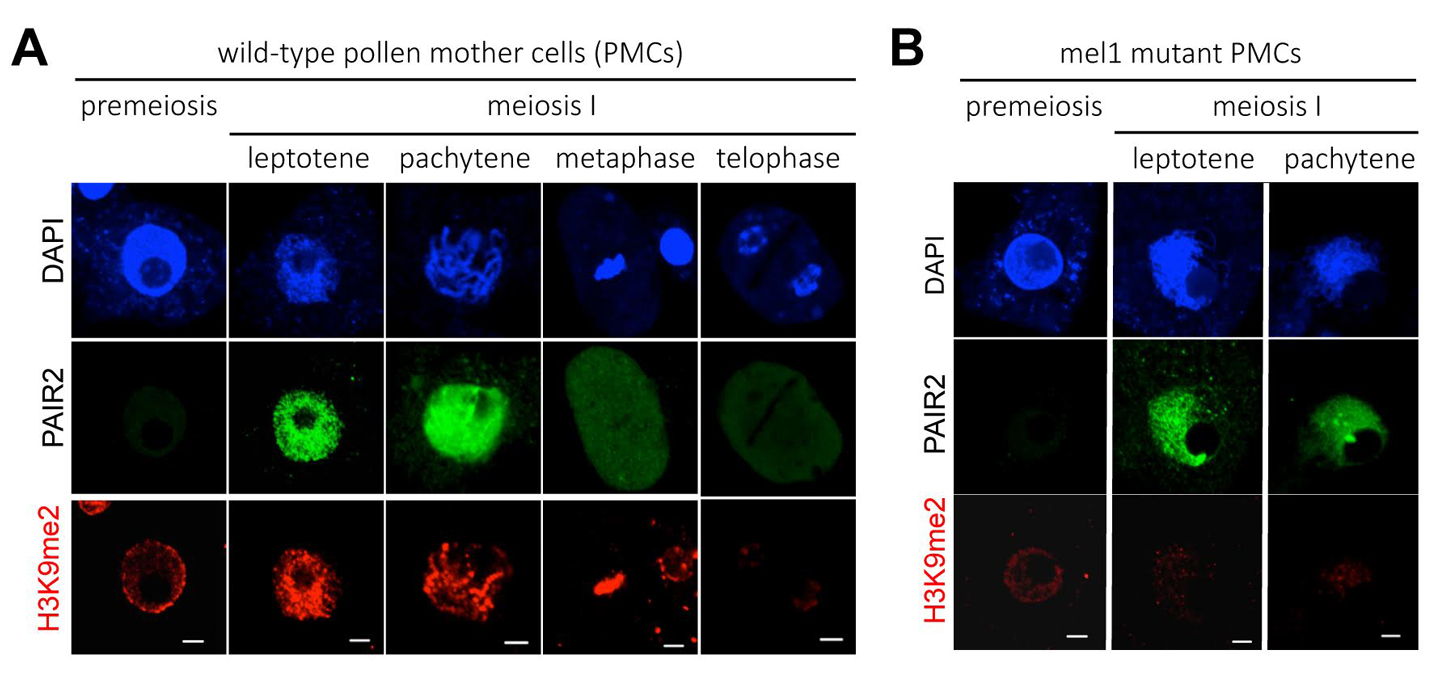
A wide reprogramming of histone H3K9me2 during meiosis I is dependent on the rice Argonaute protein MEL1.
(A) In wild-type pollen mother cells (PMCs), the level of H3K9me2 (red) is increased remarkably when cells transit from premeiosis to meiosis. Chromatin DNA is stained with DAPI (blue). PAIR2 (green) is a meiotic gene required for homologous chromosome pairing, and shows these cells undergo meiosis I. Scale bar = 5µm.
(B) mel1 mutant PMCs. PAIR2 signal (green) indicates these cells undergoing meiosis I, but no H3K9me2 reprogramming takes place.
<Glossary>
Cell Architecture Laboratory / Kimura Group
Bayesian Inference of Forces Causing Cytoplasmic Streaming in Caenorhabditis elegans Embryos and Mouse Oocytes.
Niwayama R., Nagao H., Kitajima T. S., Hufnagel L., Shinohara K., Higuchi T., Ishikawa T., Kimura A.
PLoS ONE, Vol 11, e0159917 (2016). DOI:10.1371/journal.pone.0159917
Cytoplasmic streaming is observed in wide variety of cells both in animals and plants. It is caused in many cases by cytoskeletons and molecular motors. Where these forces are exerted is difficult to identify. In this study, we developed a novel computational method to estimate the localization and amplitude of the forces generating cytoplasmic streaming. Our method infers the distribution of forces by fitting the flow field in hydrodynamics simulation to that observed in cells. We applied the method to Caenorhabditis elegans embryos and mouse oocytes. The distinct patterns of force distribution estimated in this study were consistent with the proposed distinct functions of the streaming in both of these species. We expect our method to have diverse applications, and to serve as a powerful tool for biologists who want to characterize the mechanics of biological hydrodynamic flows.
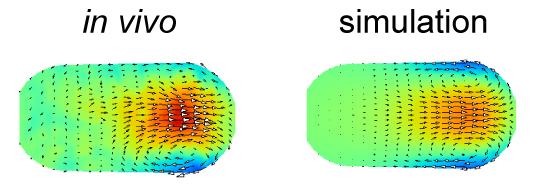
The velocity distribution in the simulation performed using the force distribution estimated in this study (right) agrees well with that measured experimentally (left). The color represents the velocity along the anterior-posterior axis, and the arrows represent the direction of the flow for cytoplasmic streaming in the C. elegans embryo.



Signing ceremony
New assistant professor joins NIG as of August 1, 2016.
Kazuo HARA: Laboratory for Gene-Expression Analysis, Okubo Group
Microbial Genetics Laboratory / Niki Group
In vitro topological loading of bacterial condensin MukB on DNA, preferentially single-stranded DNA rather than double-stranded DNA
Hironori Niki, and Koichi Yano
Scientific Reports 6, Article number: 29469 (2016) DOI:10.1038/srep29469
Condensin is the major driving force in the segregation of daughter chromosomes in prokaryotes. Core subunits of condensin belong to the SMC protein family, whose members are characterized by a unique ATPase activity and dimers with a V-shaped structure. The V-shaped dimers might close between head domains, forming a ring structure that can encircle DNA. Indeed, cohesin, which is a subfamily of SMC proteins, encircles double-stranded DNA to hold sister chromatids in eukaryotes. However, the question of whether or not condensin encircles the chromosomal DNA remains highly controversial. Here we report that MukB binds topologically to DNA in vitro, and this binding is preferentially single-stranded DNA (ssDNA) rather than double-stranded DNA. The binding of MukB to ssDNA does not require ATP. In fact, thermal energy enhances the binding. The non-SMC subunits MukF and MukE did stimulate the topological binding of MukB, although they hindered DNA-binding of MukB. Recent reports on the distribution of condensin in genomes reveal that actively transcribed genes in yeast and humans are enriched in condensin. In consideration of all these results, we propose that the binding specificity of condensin to chromosome is provided not by the DNA sequence but by the DNA structure, which is ssDNA.
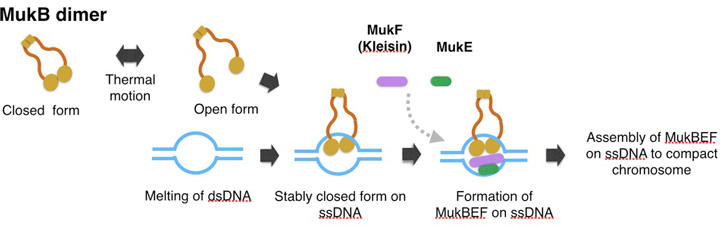
A model of topological binding of MukB in E. coli cells.
We hypothesize that the arms of MukB dimers are flexible and change between the open form or closed form depending on the thermal fluctuation. Occasionally the open form captures DNA, and then changes into the closed form. After the MukB dimer captures ssDNA, the closed form would become static because a part of the inner interface of a MukB globular domain interacts with ssDNA. Thus MukB would keep the captured DNA steady inside the ring of the dimer. Further, the MukB-DNA complex might be strengthened by MukEF, and then each of the MukBEF-DNA complexes would be assembled to compact chromosomal DNA. We infer that ATP hydrolysis is required for dissociation of the MukBEF-DNA complex from the assembled complexes.
Press release
Telomere Visualization in Tissue Sections using Pyrrole–Imidazole Polyamide Probes
Asuka Sasaki, Satoru Ide, Yusuke Kawamoto, Toshikazu Bando, Yukinori Murata, Mari Shimura, Kazuhiko Yamada, Akiyoshi Hirata, Kiyoshi Nokihara, Tatsumi Hirata, Hiroshi Sugiyama, Kazuhiro Maeshima
Scientific Reports 6: 29261 (2016) DOI:10.1038/srep29261
Press Release (Only in Japanese)
Pyrrole–Imidazole (PI) polyamides bind to specific DNA sequences in the minor groove with high affinity. Specific DNA labeling by PI polyamides does not require DNA denaturation with harsh treatments of heat and formamide and has the advantages of rapid and less disruptive processing. Previously, we developed tandem hairpin PI polyamide probes (TH59 series), which label telomeres in cultured cell lines more efficiently than conventional methods, such as fluorescence in situ hybridization (FISH). Here, we demonstrate that a TH59 derivative, HPTH59-b, along with immunostaining for specifying cell types in the tissues, visualizes telomeres in mouse and human tissue sections. Quantitative measurements of telomere length with single-cell resolution suggested shorter telomeres in the proliferating cell fractions of tumor than in non-tumor tissues. Thus, PI polyamides are a promising alternative for telomere labeling in clinical research, as well as in cell biology.
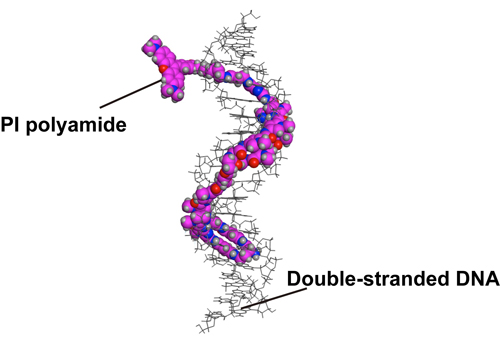
Figure1. A structural model of HPTH59-b binding to DNA.
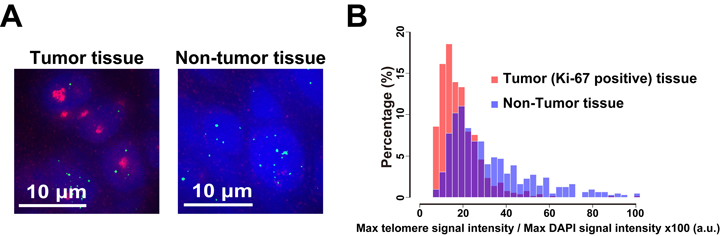
Figure2. Different telomere lengths between human tumor and non-tumor tissue sections. (A) Frozen sections of esophageal tumor/non-tumor tissue stained with DAPI (blue), anti-Ki-67 (growth marker; green) antibody, and HPTH59-b (red). Distribution histograms of telomere signal intensities in tumor and non-tumor tissue sections.
New assistant professor joins NIG as of July 1, 2016.
Daisuke TAKAO: Division of Centrosome Biology, Kitagawa Group
Division of Population Genetics / Saitou Group
Emergence and evolution of Hominidae-specific coding and noncoding genomic sequences
Morteza Mahmoudi Saber, Isaac Adeyemi Babarinde, Nilmini Hettiarachchi and Naruya Saitou
Genome Biology and Evolution Volume 8, advance access, 2016 DOI:10.1093/gbe/evw132
Family Hominidae includes humans and great apes. We analyzed whole genome sequences to find Hominidae-specific genes and highly conserved noncoding sequences (HCNSs). We discovered that Down syndrome critical region 4 (DSCR4) is the only experimentally verified gene uniquely present in Hominidae. DSCR4 has no structural homology to any known protein and was inferred to have emerged in several steps. We also identified 1,658 Hominidae-specific HCNSs. These HCNSs were found to be under purifying selection, indicating that they may harbor important functions. They are in close proximity of genes involved in sensory perception of sound and developmental process, and also showed a significantly lower nucleosome occupancy probability. Interestingly, many ancestral sequences of the Hominidae-specific HCNSs showed very high evolutionary rates. This suggests that new functions emerged through some kind of positive selection, and then purifying selection started to operate to keep these functions.
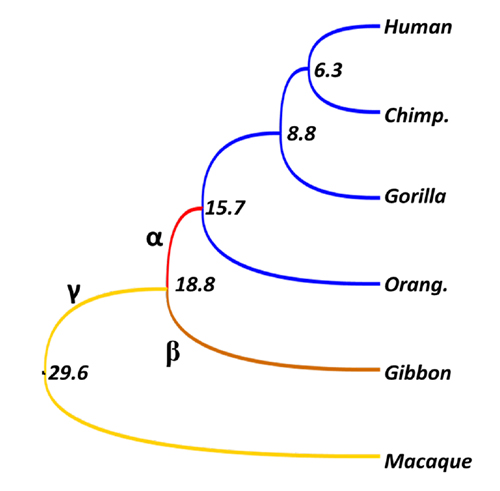
Evolutionary rates of each branch of Hominidae and their outgroups. Values are divergence times (million years). Although within-Hominidae rate is zero (identical sequences), rates for branches α, β, and γ 5.5, 2.0, and 1.9, respectively. In particular, the rate of branch α, which is common ancestor of Hominidae, is more than five times higher than neutral rate (1.0), and at least some ancestral sequences seem to experience positive selection.

Oral presentation is an integral part of scientific research and serves as an opportunity to disseminate your research to the scientific community. However scientific presentation is not an oral version of your research paper; presentation transmits not only research accomplishments but also valuable information about the presenters themselves: the breadth of their research interests, logical and critical thinking skills, future directions, and personality. To meet these hidden needs of scientific presentation we have developed a new methodology of scientific presentation, based on our experience of making many research presentations as well as attending numerous seminars. This workshop introduces essential elements of this methodology, called the “NIG Method”. NIG Method is aimed not only at nurturing skills to get your message across, but also at improving the quality of your science itself.
This two-day workshop consists of lectures and a “masterclass”. In lectures (day 1), we will discuss how research presentation differs from lectures, and describe two key structural elements of scientific presentation: “key question” and “perspective frame”. We will then introduce several techniques to aid comprehension — i.e. techniques to demonstrate your intelligence to the audience.
Masterclass session (day 2) is an opportunity to put the theory into practice. Selected participants will make a 10-minute presentation in English to receive advice from instructors and other researchers.
All activities will be conducted in English; fluency in Japanese is not required for participation.
Friday, 1 July
| 13:30-14:00: | lecture 1 | “Essence of scientific presentation” |
| 14:00-15:00: | lecture 2 | “Structure of scientific presentation” |
| 15:15-16:15: | lecture 3 | “Various techniques in scientific presentation” |
| 16:30-19:30: | Mixer | (NIG Poster Workshop) |
Saturday, 2 July
| 9:00-11:30: | Presentation masterclass | |
| 11:30-12:00: | lecture 4 | “Humor in scientific presentation” |
Researchers (Graduate Student, Postdoc, Faculty) in natural sciences
Maximum number of participants: 50
Please email the information listed below ask-ord@nig.ac.jp with the subject line: NIGmethod2016.
Participation free: None
We will be offering need-based travel grant. This grant is based on the financial need and distance required for travel to attend the workshop. To be considered, you should be registered at the time of application. Please send the information listed below with your application mail. Applicants will be notified of the results of their application by email by 21 June.
If you have any questions, write to ask-ord@nig.ac.jp
Mammalian Development Laboratory / Saga Group
GBIQ: a non-arbitrary, non-biased method for quantification of fluorescent images
Youichirou Ninomiya*, Wei Zhao and Yumiko Saga*
Scientific Reports 6, Article number: 26454 (2016) DOI:10.1038/srep26454
(* corresponding authors)
Non-arbitrary and non-biased quantification of fluorescent images is an essential tool for the data-centric approach to biological systems. Typical application is high-content analysis, where various phenotypic changes in cellular components are measured from fluorescent image data. A standard protocol to detect cellular phenotypes is cell-segmentation, in which boundaries of cellular components, such as cell nucleus and plasma membrane, are first identified to define cell segments, then acquiring various phenotypic data of each segment. To achieve reliable outcome, cell-segmentation requires manual adjustments of many parameters; this requirement could hamper automated image processing in high-throughput workflow, whose quantification must be non-arbitrary and non-biased. As a practical alternative to the method, we developed GBIQ (Grid Based Image Quantification), which allows comparison of cellular information without identification of single cells. GBIQ divides an image with tiles of fixed size grids and records statistics of the grids with their location coordinates, minimizing arbitrary intervenes. GBIQ requires only one parameter (size of grid) to be set; nonetheless it robustly produces results suitable for further statistical evaluation. The simplicity of GBIQ allows it to be readily implemented in an automated high-throughput image analysis workflow. This work was supported by the Data Assimilation and Simulation Support Technologies project from the Research Organization of Information and Systems (ROIS) in Japan.
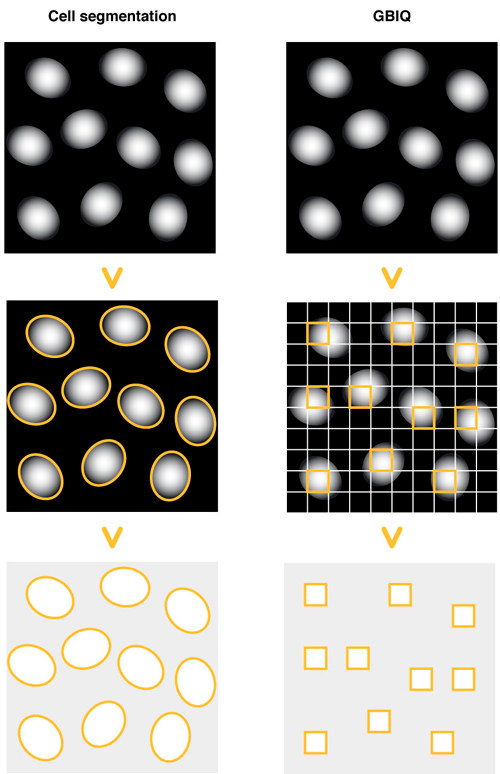
Cell segmentation: Draw outlines of cell nuclei using DNA counterstaining. Identify each outline of cell nucleus, then measure fluorescent feature of the outlines (segments).
GBIQ: Divide an image by tilling of fixed size grids. Filter certain grids that contain only cell nucleus, then measure fluorescent feature of the filtered grids.
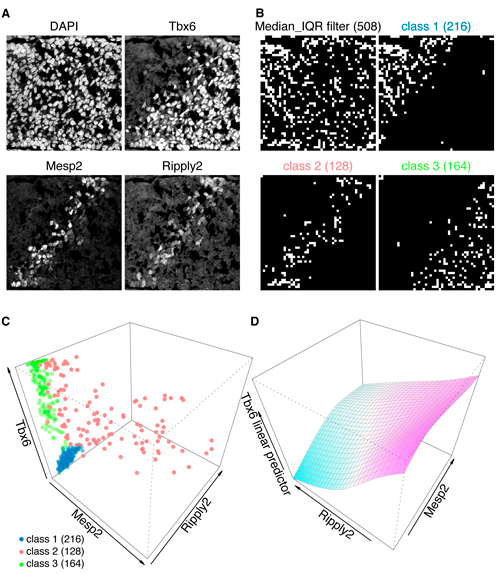
A: Triple immunofluorescent staining of developing tailbud from mouse embryo. Nuclear counterstaining by DAPI.
B and C: These immunofluorescent images (A) are processed by GBIQ with g=16. Applying the “Median_IQR filter” onto DAPI channel extracts reliable 508 observations (B, Median_IQR filter, white grids). Further applying Mclust utilizing the 3 factors (median intensities of Tbx6/Mesp2/Ripply2) onto the 508 dataset classifies them to 3 classes (C), which illustrate distinctive tissue architecture (B, class1, 2 and 3, white grids). C: All 3 factors are expressed in the class 2 (red), while neither Mesp2 nor Ripply2 expression is evident in the class 1 (blue) and class 3 (green).
D: Generalized additive model analysis of the class 2 dataset indicates negative correlation between Ripply2 and Tbx6, suggesting Ripply2 degrades Tbx6.
Biological Macromolecules Laboratory / Maeshima Group
Chromatin folding and DNA replication inhibition mediated by a highly antitumor-active tetrazolato-bridged dinuclear platinum(II) complex
Ryosuke Imai, Seiji Komeda, Mari Shimura, Sachiko Tamura, Satoshi Matsuyama, Kohei Nishimura, Ryan Rogge, Akihiro Matsunaga, Ichiro Hiratani, Hideaki Takata, Masako Uemura, Yutaka Iida, Yuko Yoshikawa, Jeffrey C. Hansen, Kazuto Yamauchi, Masato T. Kanemaki, and Kazuhiro Maeshima
Scientific Reports 6, Article number: 24712 (2016) DOI:10.1038/srep24712
Chromatin DNA must be read out for various cellular functions, and copied for the next cell division. These processes are targets of many anticancer agents. Platinum-based drugs, such as cisplatin, have been used extensively in cancer chemotherapy. The drug–DNA interaction causes DNA crosslinks and subsequent cytotoxicity. Recently, it was reported that an azolato-bridged dinuclear platinum (II) complex, 5-H-Y, exhibits a different anticancer spectrum from cisplatin. Here, using an interdisciplinary approach, we reveal that the cytotoxic mechanism of 5-H-Y is distinct from that of cisplatin. 5-H-Y inhibits DNA replication and also RNA transcription, arresting cells in the S/G2 phase, and are effective to cisplatin-resistant cancer cells. Moreover, it causes much less DNA crosslinking than cisplatin, and induces chromatin folding. 5-H-Y will expand the clinical applications for the treatment of chemotherapy-insensitive cancers.
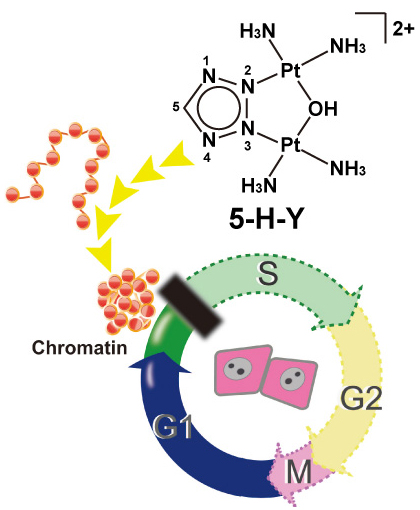
5-H-Y inhibits DNA replication and arrests the treated cells in S/G2 phase. 5-H-Y binds tightly to chromatin DNA and induces chromatin folding in vitro and in vivo.
Division of Population Genetics / Saitou Group
Genomic locations of conserved noncoding sequences and their proximal protein-coding genes in mammalian expression dynamics
Isaac Adeyemi Babarinde and Naruya Saitou
Molecular Biology and Evolution DOI:10.1093/molbev/msw058
Conserved Noncoding Sequences (CNSs) in mammals were examined with special reference to their genomic location. We extracted the CNSs conserved between chicken and four mammalian species (human, mouse, dog and cattle). Human CNSs were confirmed to be under purifying selection. The distribution pattern, ChIP-Seq and RNA-Seq data suggested that the CNSs are regulatory elements. Physical distances between CNS and their nearest protein coding genes were well conserved between human and mouse genomes. ChIP-Seq signal and gene expression patterns also suggested that CNSs regulate nearby genes. Genes with more CNSs have more evolutionarily conserved expression than those with fewer CNSs. These results suggest that the genomic locations of CNSs are important for their regulatory functions. In fact, various kinds of evolutionary constraints may be acting to maintain the genomic locations of CNSs and protein-coding genes in mammals to ensure proper regulation. First author of this paper, Dr. Babarinde, just receive Ph.D. from Department of Genetics, SOKENDAI. He also received Morishima Award from NIG.
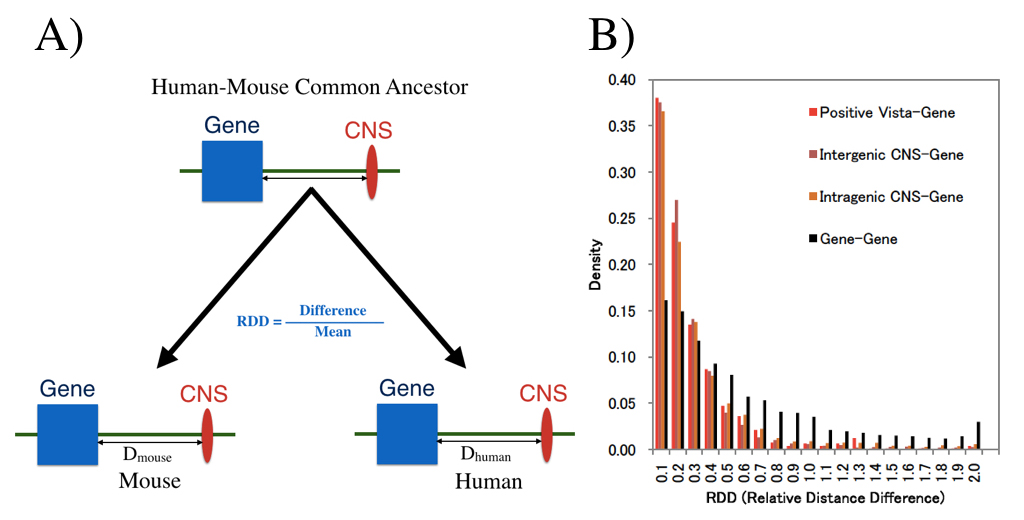
We examined whether genomic distances between genes and CNSs which already existed in the common ancestor of human and house are conserved now. (A) shows definition of distance measure RDD, and (B) shows RDD distribution. Distances between CNS and genes are much more conserved than those between genes.










