



![]()
Latent environment allocation of microbial community data
Koichi Higashi, Shinya Suzuki, Shin Kurosawa, Hiroshi Mori and Ken Kurokawa
PLOS Computational Biology Published: June 6, 2018 DOI:10.1371/journal.pcbi.1006143
Pressrelease (In Japanese only)
As data for microbial community structures found in various environments has increased, studies have examined the relationship between environmental labels given to retrieved microbial samples and their community structures. However, because environments continuously change over time and space, mixed states of some environments and its effects on community formation should be considered, instead of evaluating effects of discrete environmental categories. Here we applied a hierarchical Bayesian model to paired datasets containing more than 30,000 samples of microbial community structures and sample description documents. From the training results, we extracted latent environmental topics that associate co-occurring microbes with co-occurring word sets among samples. Topics are the core elements of environmental mixtures and the visualization of topic-based samples clarifies the connections of various environments. Based on the model training results, we developed a web application, LEA (Latent Environment Allocation), which provides the way to evaluate typicality and heterogeneity of microbial communities in newly obtained samples without confining environmental categories to be compared. Because topics link words and microbes, LEA also enables to search samples semantically related to the query out of 30,000 microbiome samples.
Source: Koichi Higashi, et al., (2018), 14,e1006143, PLOS Computational Biology, DOI:10.1371/journal.pcbi.1006143
 |
 |
| College of Life Science, National Taiwan University (JENG, Shih-Tong, Dean and TING, Chau-Ti, Associate Professor) |
National Institute of Genetics (KATSURA, Isao, Director-General and KAWAKAMI, Koichi, Professor) |
Requirement of the 3′-UTR-dependent suppression of DAZL in oocytes for pre-implantation mouse development
Kurumi Fukuda, Aki Masuda, Takuma Naka, Atsushi Suzuki, Yuzuru Kato, and Yumiko Saga
Plos Genetics Published: June 8, 2018 DOI:10.1371/journal.pgen.1007436
Oogenesis is regulated by precise gene expression. One of important gene of mouse oogenesis is Dazl which has a role in translation promotion and indispensable for embryonic oocytes. Dazl is thought to be important whole life of oocyte because the expression of Dazl mRNA is detectable from embryonic to postnatal stage. In this study, we found that Dazl protein need to be suppressed in postnatal ovary whereas it has indispensable role in embryonic ovary. If this regulation does not work, female causes litter size reduction due to the defect in pre-implantation development. Thus, switching the Dazl expression from embryonic to postnatal stage by post-transcriptional regulation via Dazl’s 3’UTR is crucial for regulation production of next generation.
This study was partly supported by Grant-in-Aid for Young Scientists (B) to Y.K. (No. 25840091) and Grant-in-Aid for Scientific Research (A) to Y.S. (No. 26251025) from JSPS and by Grant-in-Aid for Scientific Research on Innovative Areas from MEXT to Y.S. (No. 25112002), A.S. (No. 16H01252), and Y.K. (No. 16H01259). K.F. is a JSPS Research Fellow (No. 16J11687).
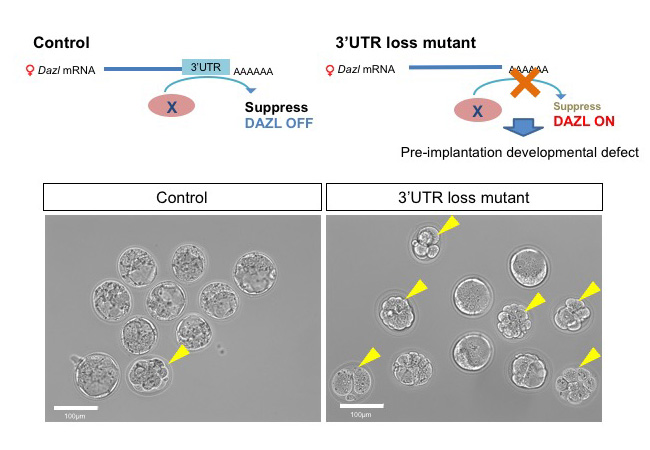
Figure: DAZL expression is suppressed in postnatal ovary in control (Upper left scheme). On the other hands, the mutant oocytes which lack 3’UTR of Dazl retain DAZL expression even in the postnatal stage and exhibit defects during preimplantation zygote stages (Upper right scheme). In control, most of all E3.5 zygotes became blastocysts (lower left panel), but in the mutant half of them stopped development (lower right panel). Yellow arrow heads indicate zygotes showing arrested development.
VITCOMIC2: visualization tool for the phylogenetic composition of microbial communities based on 16S rRNA gene amplicons and metagenomic shotgun sequencing.
Mori H, Maruyama T, Yano M, Yamada T, Kurokawa K. BMC Syst Biol
BMC Systems Biology 12 (Suppl 2), 30, 2018. DOI:10.1186/s12918-018-0545-2
Dr. Hiroshi Mori and Dr. Ken Kurokawa in the Genome Evolution laboratory from the Center for Information Biology in National Institute of Genetics, and colleagues developed a web application “VITCOMIC2” to infer taxonomic composition of a microbial community from the metagenomic sequencing data and the 16S rRNA gene amplicon sequencing data. VITCOMIC2 conducts fast and accurate taxonomic assignment of metagenomic data by using a GPU-based DNA sequence similarity search tool CLAST. Using VITCOMIC2, users can avoid the taxonomic assignment ambiguities which are derived from sequence clustering before conducting taxonomic assignment. VITCOMIC2 can be used in http://vitcomic.org .
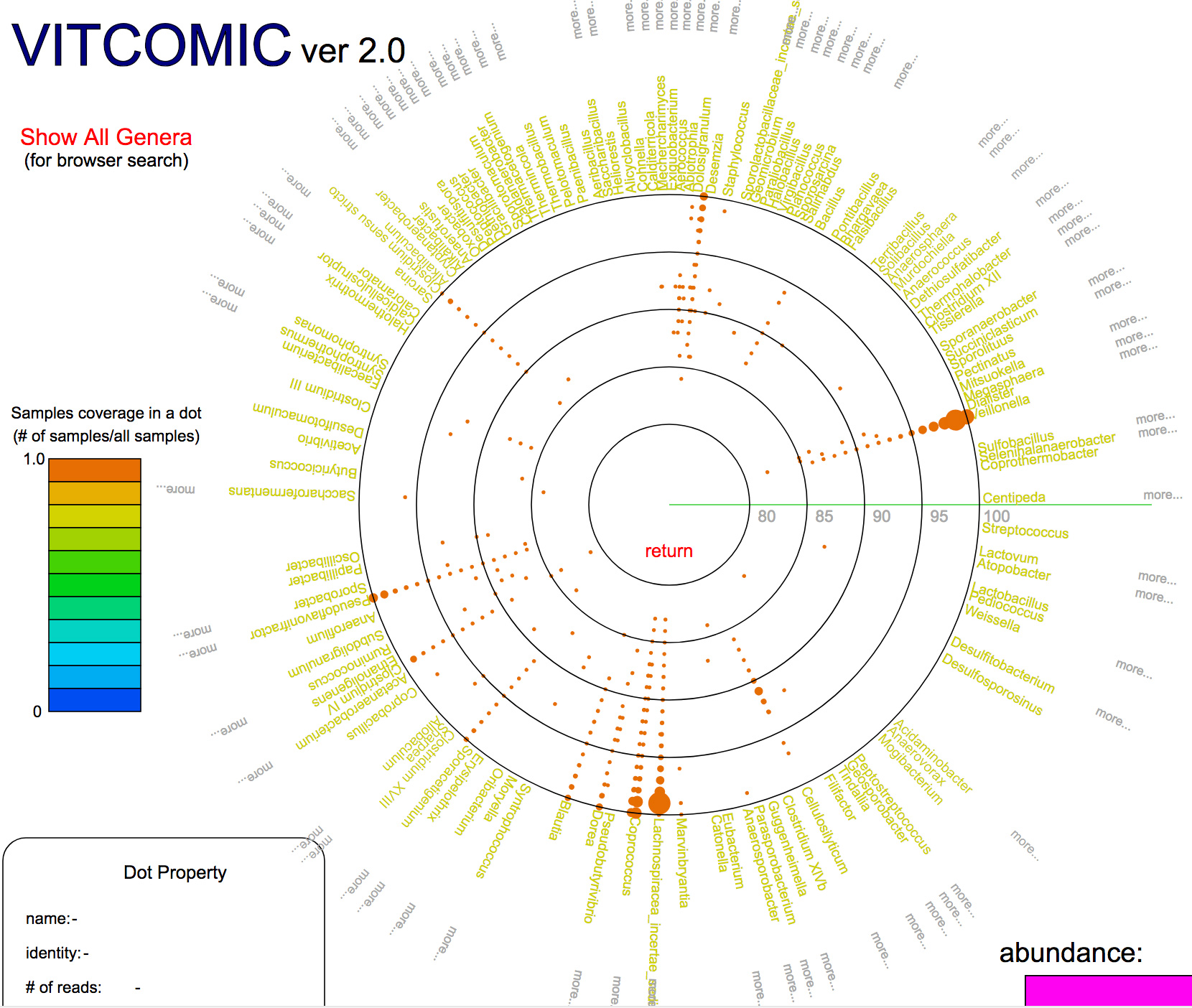
図:Example of a VITCOMIC2 result
New assistant professor joins NIG as of June 1, 2018.
Takayuki TORISAWA: Cell Architecture Laboratory • Kimura Group
![]()
Identification of a neuronal population in the telencephalon essential for fear conditioning in zebrafish
Pradeep Lal, Hideyuki Tanabe, Maximiliano L. Suster, Deepak Ailani, Yuri Kotani, Akira Muto, Mari Itoh, Miki Iwasaki, Hironori Wada, Emre Yaksi , and Koichi Kawakami
BMC Biology Published: 25 April 2018 DOI:10.1186/s12915-018-0502-y
EurekAlert! link about this artcle
Have you ever wondered why animals avoid dangers by sensing some “signs” possibly related to the danger? A simple form of this phenomenon is called “fear conditioning”, which is a type of learning commonly seen in every animal on the earth. By manipulating activity of specific neurons of the zebrafish brain, scientists at the National Institute of Genetics (NIG) in Japan have elucidated a neuronal population essential for fear conditioning in zebrafish. The study, published in the April 25 issue of BMC Biology, suggests that such a neural circuit essential for fear conditioning exists and is conserved during vertebrate evolution.
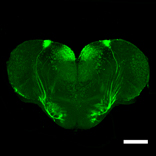
Figure: A section of the zebrafish telencephalon. The neurons essential for fear conditioning are illuminated with GFP (green fluorescence protein). Scale bars: 200 μm.
Video 1: Fear conditioning of zebrafish. The fish was placed in a plastic box with two compartments. 10 seconds after LED was on, an electric shock was given (day 1). This was repeated 10 times a day for five consecutive days. On day 5, when LED was on, the fish escaped to another compartment.
Video 2: 3D image of the neurons essential for fear conditioning. Transparent brain was created and analyzed by light-sheet microscopy.
Epidermal regulation of bone morphogenesis through the development and regeneration of osteoblasts in the zebrafish scale.
Iwasaki M., Kuroda J., Kawakami K., Wada H.
Developmental Biology 437, 105-119, 2018. DOI:10.1016/j.ydbio.2018.03.005
The scale of the teleost fish is a dermal bone embedded in the skin. We investigated the mechanisms of bone patterning in the zebrafish scale by using transgenic lines that visualize osteoblasts. We showed that the zebrafish scale contains two distinct types of osteoblasts: a monolayer sheet of central osteoblasts along the surface of scales; and marginal osteoblasts elongated along the scale edge (Fig. A). During scale growth, central osteoblasts progressively increase in size without cell proliferation. Sonic hedgehog (shh) is expressed in the epidermal cells overlying marginal osteoblasts. Inhibition of Hh signaling reduces the number of marginal osteoblasts and interferes with scale growth. Moreover, inhibition of Wnt/planar cell polarity (PCP) signaling in the epidermis caused a misorientation of scales (Fig. B), correlated to the altered expression pattern of shh. This study reveals a novel role of the epidermis in the regulation of bone patterning.
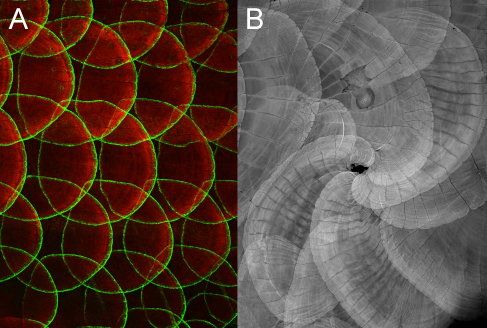
Figure: (A) Gal4 enhancer trap line (hspGFFDMC13F;UAS:GFP) expressing GFP in a specific population of osteoblasts along the scale edge. (B) Disrupted Wnt/PCP signaling in the epidermis causes a misorientation of scales, indicating a role of the epidermis in bone patterning.
Wilms Tumor 1b defines a wound-specific sheath cell subpopulation associated with notochord repair
Lopez-Baez, J.C., Simpson, D.J., Forero, L.L., Zeng, Z., Brunsdon, H., Salzano, A., Brombin, A., Wyatt, C., Rybski, W., Huitema, L.FA., Dale, R.M., Kawakami, K., Englert, C., Chandra, T., Schulte-Merker, S., Hastie, N.D., and Patton, E.E.
eLife 2018;7:e30657 DOI:10.7554/eLife.30657
Regenerative therapy for degenerative spine disorders requires the identification of cells that can slow down and possibly reverse degenerative processes. Here, we identify an unanticipated wound-specific notochord sheath cell subpopulation that expresses Wilms Tumor (WT) 1b following injury in zebrafish. We show that localized damage leads to Wt1b expression in sheath cells, and that wt1b+cells migrate into the wound to form a stopper-like structure, likely to maintain structural integrity. Wt1b+sheath cells are distinct in expressing cartilage and vacuolar genes, and in repressing a Wt1b-p53 transcriptional programme. At the wound, wt1b+and entpd5+ cells constitute separate, tightly-associated subpopulations. Surprisingly, wt1b expression at the site of injury is maintained even into adult stages in developing vertebrae, which form in an untypical manner via a cartilage intermediate. Given that notochord cells are retained in adult intervertebral discs, the identification of novel subpopulations may have important implications for regenerative spine disorder treatments.
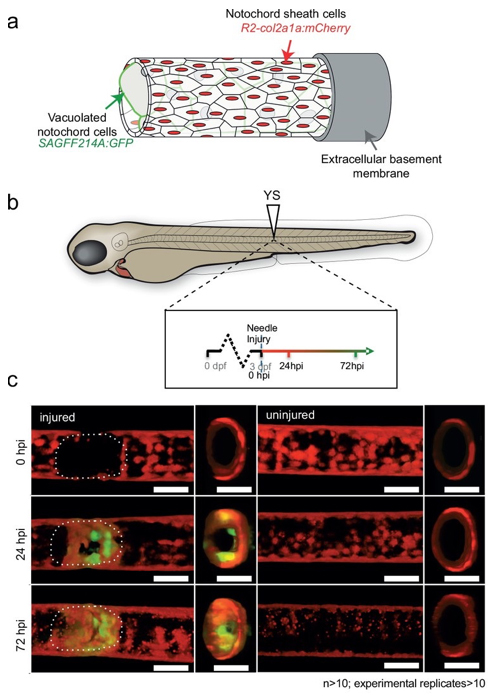
Figure: In transgenic fish expressing GFP in the notochord vacuolated cells, GFP-expressing cells appeared 24-72 hours after injury.
A novel zebrafish intestinal tumor model reveals a role for cyp7a1-dependent tumor-liver crosstalk in tumor’s adverse effects on host
Sora Enya, Koichi Kawakami, Yutaka Suzuki, Shinpei Kawaoka
Disease Models & Mechanisms (2018) DOI:10.1242/dmm.032383
The nature of host organs and genes that underlie tumor-induced physiological disruption on host has been poorly understood. Here, we establish a novel zebrafish intestinal tumor model, and find that hepatic cyp7a1, the rate-limiting factor for synthesizing bile acids (bile alcohol (BA) in zebrafish) is important for such a phenomenon. We created a transgenic zebrafish line, in which an oncogenic form of kras (krasG12D) was expressed in the posterior intestine by the Gal4-UAS method, and found that the intestinal tumor was formed. The intestinal tumor then caused detrimental effects on host, including liver inflammation, hepatomegaly, growth defects, and organismal death. Whole-organismal level gene expression analysis and metabolite measurements revealed that the intestinal tumor reduced total BA levels possibly via altered expression of hepatic cyp7a1. We demonstrated overexpression of cyp7a1 in the liver restored the BA synthesis and ameliorated tumor-induced liver inflammation. Thus, we discovered a previously unknown role of cyp7a1 as the host gene that links the intestinal tumor to the hepatic cholesterol-BA metabolism and liver inflammation. Our model provides an important basis to investigate host genes responsible for tumor-induced phenotypes and to uncover mechanisms underlying how tumors adversely affect host organisms.
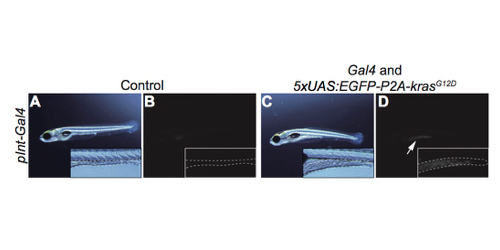
Figure: A transgenic zebrafish model bearing the intestine tumor created by the Gal4-UAS method. A, B; control. C, D: transgenic fish. B, D: fluorescent images. Dotted lines indicate the shape and size of the intestine. An arrow indicates EGFP expression.
Maser: one-stop platform for NGS big data from analysis to visualization
Sonoko Kinjo, Norikazu Monma, Sadahiko Misu, Norikazu Kitamura, Junichi Imoto, Kazutoshi Yoshitake, Takashi Gojobori, Kazuho Ikeo
Database (2018) Vol. 2018: article ID bay027; DOI:10.1093/database/bay027
A major challenge in analyzing the data from high-throughput next-generation sequencing (NGS) is how to handle the huge amounts of data and variety of NGS tools and visualize the resultant outputs. To address these issues, we developed a cloud-based data analysis platform, Maser (Management and Analysis System for Enormous Reads), and an original genome browser, Genome Explorer (GE).
Maser realized a more user-friendly analysis platform especially for beginners by improving graphical display and providing the selected standard pipelines that work with built-in genome browser. All the analyses executed on Maser are recorded in the analysis history, helping users to trace and repeat the analyses. The entire process of analysis and its histories can be shared with collaborators or opened to the public. In conclusion, our system is useful for managing, analyzing, and visualizing NGS data and achieves traceability, reproducibility, and transparency of NGS analysis.
Maser is currently supported by Japan Agency for Medical Research and Development (AMED).
Figure 1: Maser tutorial and user registration page
Figure 2: Maser’s Top Page (Only registered user can access)
We set up a research group called “Rinkai Hack” with a few volunteers to promote research education and human resources exchanges beyond the boundaries of informatics and animal/ plant research, with marine stations as the primary places of activity. The name Rinkai Hackathon is derived from “Rinkai (coastal, waterfront, or marine), Hack, marathon”.
We widely invite for participants of the event of this year, “RinkaiHackathon 2018”.We will hold a symposium on June 10and hands-on, three-day course June 11-13.The venue of the symposium is Fukuju Kaikan in Fukuyama Castle, and the venue of the hands-on course is the Mukai-Shima marine station of Hiroshima University (Mukaijima, Onomichi, Hiroshima Prefecture).In addition to lectures by the committee members, several invited speakers are scheduled. For details, please see the event page.
https://sites.google.com/view/rinkaihack/
This year we are planning lectures and practical training on environmental DNA / metagenome and will provide experience-based learnings from DNA extraction from seawater to DNA sequence analysis. Let’s learn the actual latest situation of the marine metagenomic analyses which is often taking up as a scientific topic recently. At the same time, we will set a discussion time in which participants discuss what of classical knowledge and system of biology does not change depending on the progress of technology.
As of this year, we will change the primary language during the event to English, because we want to encourage domestic and international students to participate and also have opportunities to exchange outside the university. At the same time, we also encourage not only Japanese students who are confident in English but also students who are not as confident. If you cannot communicate well in English, the organizing staff will support you. (The committee members are also not very good at English. But we are doing it, somehow!! Que Sera Sera.)
For details of the event, please visit the website.
https://sites.google.com/view/rinkaihack/registration
Registration is accepted from the registration form on the website.
https://sites.google.com/view/rinkaihack/coming-events/rinkai-hackathon-2018
Any question related to the event is also accepted from the website.
https://sites.google.com/view/rinkaihack/coming-events/rinkai-hackathon-2018/moredetailsrinkaihackathon2018?authuser=0
Intended participants: graduate students and postdoctoral fellows.
![]()
Division of Neurogenetics
Mbf1 ensures Polycomb silencing by protecting E(z) mRNA from degradation by Pacman
Kenichi Nishioka, Xian-Feng Wang, Hitomi Miyazaki, Hidenobu Soejima, Susumu Hirose
Development 2018 145:dev162461 DOI:10.1242/dev.162461
Pressrelease (In Japanese only)
Under stress conditions, the coactivator Multiprotein-bridging factor 1 (Mbf1) translocates from the cytoplasm into the nucleus to induce stress-response genes. However, its role in the cytoplasm, where it is mainly located, has remained elusive. Here, we show that Drosophila Mbf1 associates with E(z) mRNA and protects it from degradation by the exoribonuclease Pacman, thereby ensuring Polycomb silencing. This mechanism would also allow flexibility in Polycomb silencing, as Mbf1 protein expression declines upon differentiation.
In addition to E(z) mRNA, Mbf1 binds to mRNAs of various stress-response genes. Therefore, Mbf1 appears to contribute to various types of stress defense as both a nuclear coactivator and as a cytoplasmic mRNA-stabilizing protein. It is intriguing that Mbf1 contributes to the same biological function through different subcellular localisation.
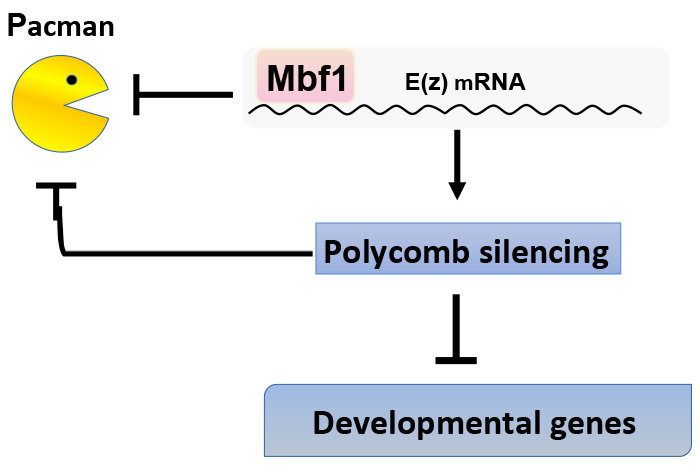
Mbf1 binds to E(z) mRNA and protects it from degradation by Pacman. Polycomb silencing represses expression of the pacman gene. Therefore, Mbf1 ensures robust Polycomb silencing.
Construction of a simple evaluation system for the intestinal absorption of an orally administered medicine using Bombyx mori larvae
Fumika Ichino, Hidemasa Bono, Takeru Nakazato, Atsushi Toyoda, Asao Fujiyama, Kikuo Iwabuchi, Ryoichi Sato, Hiroko Tabunoki
Drug Discoveries & Therapeutics, 12(1) 7-15, 2018 DOI:10.5582/ddt.2018.01004
Human intestinal absorption is estimated using a human colon carcinoma cell line (Caco-2) cells from human colorectal adenocarcinoma, intestinal perfusion, or a mammalian model. These current evaluation systems are limited in their ability to estimate human intestinal absorption. In addition, in vivo evaluation systems using laboratory animals such as mice and rats entail animal ethics problems, and it is difficult to screen compounds on a large scale at the drug discovery stage. Thus, we propose the use of Bombyx mori larvae for evaluation of intestinal absorption of compounds as an alternative system in this study.
Prof. Atsushi Toyoda and Prof. Asao Fujiyama (Center for Information Biology) contributed to sequencing midgut transcriptome of B. mori. Dr. Hidemasa Bono and Dr. Takeru Nakazato (Database Center for Life Science) contributed to this work in comparative analysis of Caco-2 cells, human and B.mori gut transcriptomes.
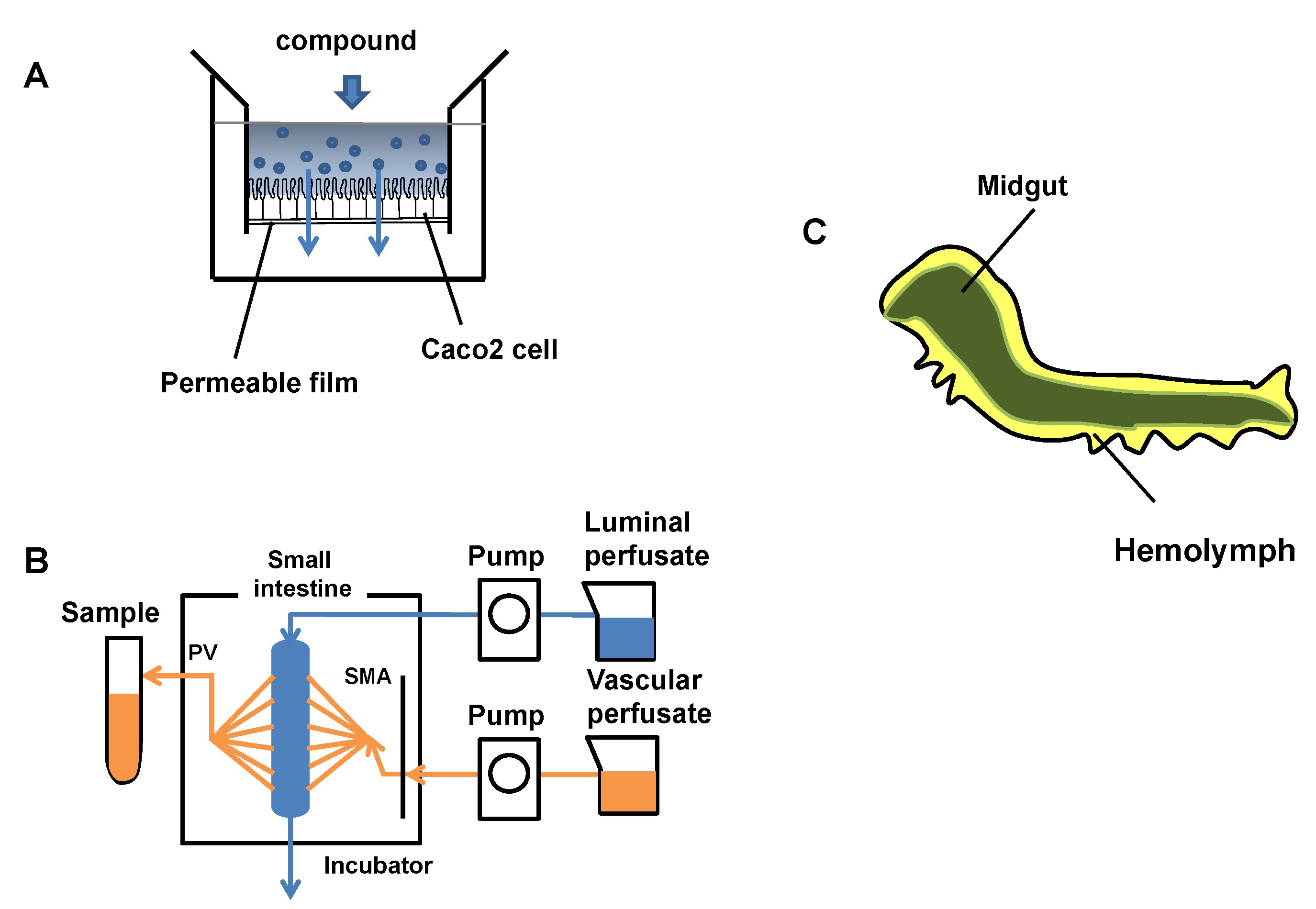
Figure: Evaluation of model systems for intestinal permeability and the internal structure of B.mori larva. Most of the body is occupied by the midgut (green), and its surroundings are filled with hemolymph(yellow).
Collaborative environmental DNA sampling from petal surfaces of flowering cherry Cerasus × yedoensis ‘Somei-yoshino’ across the Japanese archipelago
Ohta T, Kawashima T, Shinozaki NO, Dobashi A, Hiraoka S, Hoshino T, Kanno K, Kataoka T, Kawashima S, Matsui M, Nemoto W, Nishijima S, Suganuma N, Suzuki H, Taguchi Y, Takenaka Y, Tanigawa Y, Tsuneyoshi M, Yoshitake K, Sato Y, Yamashita R, Arakawa K, Iwasaki W
Journal of Plant Research 2018, Epub ahead of print. DOI:10.1007/s10265-018-1017-x
A collaborative research project “The Ohanami project” was launched in 2015 by the organizers of the conference of high-throughput sequencing technologies, the NGS Field 4th meeting. The project aimed to collect environmental DNA (eDNA) from petal surfaces of flowering cherry Cerasus × yedoensis ‘Somei-yoshino’ to analyze the origins of eDNA found on the plants. Somei-yoshino is a cultivar widely cultivated across the Japanese archipelago, and the trees are all clones of a single tree since it is propagated through grafting and self-incompatible. Over 150 collaborators joined the sampling campaign of the project to collect eDNA samples from 577 locations (Fig. 1). The project team performed 16S rRNA amplicon sequencing method to analyze the origins of eDNA. As a result of DNA sequencing, we found that the DNA sequences of common plants including the one like Japanese Ceder are found on the petal surfaces along with the DNA of flowering cherry itself. This project is the first project that revealed the existence of eDNA on petal surfaces with many samples from a broad geographical range. The project also showed that a crowd-sourcing approach is applicable on DNA sampling from a wide range of locations in a short term.

Fig. 1: DNA sampling from petal surfaces of flowering cherry. Collaborators used the swab kit to collect environmental DNA from the petal surfaces.
New assistant professor joins NIG as of March 1, 2018.
Keiko TAKANAMI: Mouse Genomics Resource Laboratory (MGRL), Koide Group
Experimental Farm / Nonomura Group
EAT1 transcription factor, a non-cell-autonomous regulator of pollen production, activates meiotic small RNA biogenesis in rice anther tapetum.
Seijiro Ono, Hua Liu, Katsutoshi Tsuda, Eigo Fukai, Keisuke Tanaka, Takuji Sasaki, Ken-Ichi Nonomura.
PLOS Genetics, 14 (2), e1007238, (2018) DOI:10.1371/journal.pgen.1007238
Small RNA plays important roles in development, anti-viral defense and etc., mediated by transcriptional and post-transcriptional of repression of gene expressions. Here, we revealed that the EAT1 transcription factor triggers the biosynthesis of 24-nt small RNAs during meiosis in rice anther tapetum. EAT1 promoted the transcription of long precursor RNAs, as small RNA precursors, and DCL5 gene, required for small RNA processing (Fig. 1).
This study demonstrated a possibility that a subset of EAT1-dependent 24-nt phasiRNAs are transferred from tapetal cells to adjoining meiocytes, from the analysis of MEL1, the Argonaute protein specifically expressed in meiocytes (Fig. 1C). This finding strongly suggests that the reproductive phasiRNAs may act as a mediator of orchestrated development of germ cells and surrounding somatic cells.
This work is a collaboration with Tokyo University of Agriculture and Niigata University.
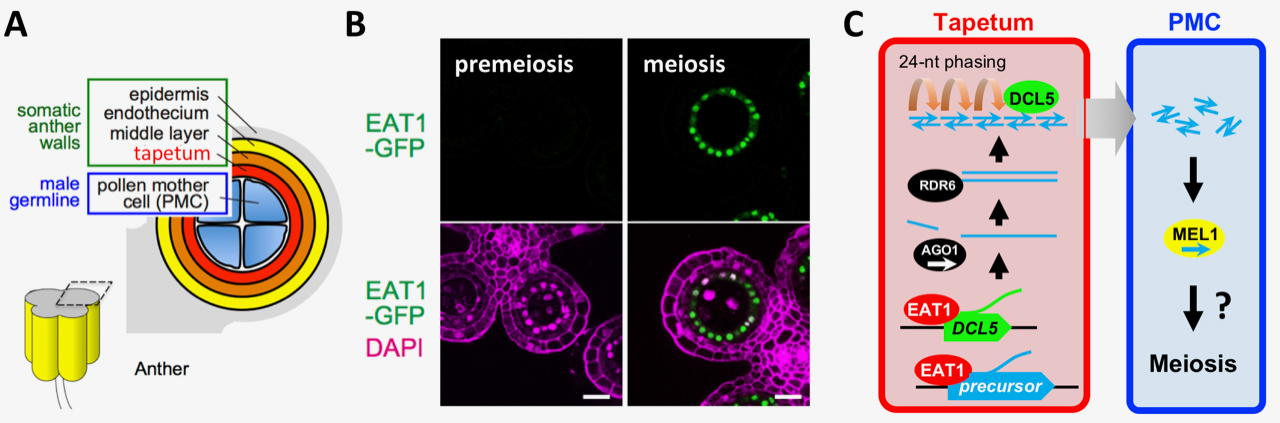
Fig. 1. EAT1 transcription factor promotes the biosynthesis of 24-nt meiotic small RNA (sRNA) in rice anther tapetum
(A) A cross section of an anther lobe. Tapetum is adjoining to male meiocytes (PMCs). (B) EAT1 (green) accumulates in tapetal cell nuclei at meiosis (right), but not at premeiosis (left). Bar=20µm. (C) EAT1 activates transcription of sRNA precursors and DCL5 gene in tapetum. After primary processing and double-strandization, precursor RNAs are sliced into 24 nt by DCL5. MEL1 binding to a subset of EAT1-dependent sRNA suggests intercellular mobilization of meiotic sRNA.










