



Mammalian Development Laboratory / Saga Group
Dead end1 is an essential partner of NANOS2 for selective binding of target RNAs in male germ cell development
Atsushi Suzuki, Yuki Niimi, Kaori Shinmyozu, Zhi Zhou, Makoto Kiso and Yumiko Saga
EMBO report. 2015 DOI:10.15252/embr.201540828
Mammalian Development RNA-binding proteins (RBPs) play important roles for generating various cell types in many developmental processes, including eggs and sperms. Nanos is widely known as an evolutionarily conserved RNA-binding protein implicated in germ cell development. Mouse NANOS2 interacts directly with the CCR4-NOT (CNOT) deadenylase complex, resulting in the suppression of specific RNAs. However, the mechanisms involved in target specificity remain elusive. We show that another RBP, Dead end1 (DND1), directly interacts with NANOS2 to load unique RNAs into the CNOT complex. This interaction is mediated by the zinc finger domain of NANOS2, which is essential for its association with target RNAs. In addition, the conditional deletion of DND1 causes the disruption of male germ cell differentiation similar to that observed in Nanos2-KO mice. Thus, DND1 is an essential partner for NANOS2 that leads to the degradation of specific RNAs. This research is partly supported by Grant-in-Aid for Scientific Research on Innovative Areas ”Epigenome dynamics and regulation in germ cells” to YS and “Mechanism regulating gamete formation” to AS.
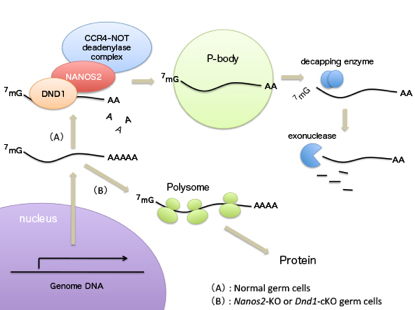
Nanos2 directly interacts with DND1, by which target RNAs are recruited to P-body for degradation. This mechanism is essential for male germ cell development.
Experimental Farm • Nonomura Group
Rice MEL2, the RNA recognition motif (RRM) protein, binds in vitro to meiosis-expressed genes containing U-rich RNA consensus sequences in the 3′-UTR
Saori Miyazaki, Yutaka Sato, Tomoya Asano, Yoshiaki Nagamura, Ken-Ichi Nonomura
Plant Molecular Biology, Published online. 30 August, 2015. DOI:10.1007/s11103-015-0369-z
Developmental processes of plant germline are strictly regulated in angiosperms, to complete seed production during a short limited season. Synchronous male meiosis within a same anther, resulting in simultaneous production of pollen grains, is an example. Our group previously identified a rice protein, MEL2, regulating proper timing of meiosis transition. However, the mechanisms are largely ambiguous yet.
This paper identified RNA consensus sequences that MEL2 preferentially binds in vitro. Searched for the rice genome, the consensus-like sequences were frequently found in 3′-untranslated regions (3′-UTR) of 249 genes. Proteomic analyses revealed that several proteins that altered their amounts in mel2 mutant anthers were supposed to function in meiosis, and that the 3′-UTRs of their corresponding mRNAs contained MEL2-binding consensus-like sequences. These results suggest that MEL2 regulates meiosis transition via translational control of meiotic target mRNAs.
This is a collaborative work with National Institute of Agrobiological Sciences (NIAS) and Kanazawa University. This study was supported by JSPS KAKENHI and JSPS Bilateral Program.
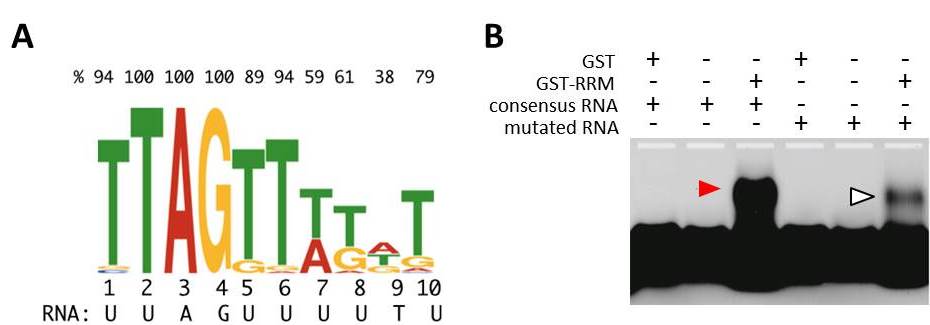
MEL2 protein binds uridine (U)-rich RNA sequences in vitro.
(A) 10-nucleotides RNA consensus sequence that RNA recognition motif (RRM) of MEL2 preferentially binds in vitro. Top values indicate the percentage of a prior residue (T, A, G or C) in each position.
(B) Electrophoresis mobility shift assay. The RNA-band position was shifted only when MEL2-RRM was mixed with consensus RNA sequence labeled with the fluorescense (red arrowhead). Mobility was significantly affected in the mutation-induced consensus (open arrowhead).
Division of Population Genetics / Saitou Group
Unique characteristics of the Ainu population in Northern Japan.
Jinam Timothy A, Kanzawa-Kiriyama Hideaki, Inoue Ituro, Tokunaga Katsushi, Omoto Keiichi, and Saitou Naruya.
Journal of Human Genetics. 2015 Jul 16. DOI:10.1038/jhg.2015.79
There are three human populations inhabiting the Japanese Archipelago: the Ainu, mostly living in Hokkaido island in the north; Ryukyuans who mostly reside in the Ryukyu islands in the south; and Mainland Japanese who can be found throughout the Japanese Archipelago. By analyzing up to 600 thousand genome-wide autosomal Single Nucleotide Polymorphisms (SNP) in these populations, we, in collaboration with the University of Tokyo, found that highly differentiated SNPs between the Ainu and the Mainland Japanese are associated with genes (EDAR and COL7A1) related to hair & tooth and facial morphology, respectively (Fig. A). Assuming the Ainu as descendants of the Jomon people (hunter-gatherers) and continental Asians (Han Chinese and Koreans) as descendants of migrant people (farmers) (Fig. B), the proportion of Jomon genetic component in Mainland Japanese was ~20%. The admixture event was estimated to have occurred at least 55 generations ago, or 7th Century. These results clearly demonstrate the unique phylogenetic status of the Ainu people within East Asia. This study was supported by SOKENDAI strategic interdisciplinary research. First author, Timothy A. Jinam, is assistant professor of Saitou Laboratory, coauthor Hideaki Kanzawa-Kiriyama graduated SOKENDAI Department of Genetics in 2014, and Ituro Inoue is professor of Division of Human Genetics at this Institute.
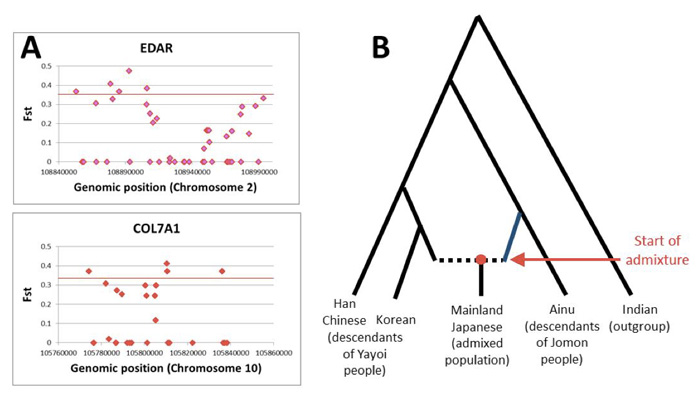
A) Some SNPs residing in two genes related to hair & tooth and facial morphology show large frequency differences between the Ainu and the Mainland Japanese.
B) The admixture model of the Mainland Japanese.
Mammalian Development Laboratory / Saga Group
The RNA binding protein Nanos2 organizes a post-transcriptional buffering system to retain primitive states of mouse spermatogonial stem cells
Zhi Zhou, Takayuki Shirakawa, Kazuyuki Ohbo, Aiko Sada, Wu Quan, Kazuteru Hasegawa, Rie Saba, Yumiko Saga Developmental Cell. Published Online: June 25, 2015 DOI:http://dx.doi.org/10.1016/j.devcel.2015.05.014In many adult tissues, homeostasis relies on self-renewing stem cells that are primed for differentiation. The reconciliation mechanisms of these characteristics remain a fundamental question in stem cell biology. Here, we show that Nanos2, an evolutionarily conserved RNA-binding protein, works with other cellular messenger ribonucleoprotein (mRNP) components to ensure the primitive status of SSCs through a dual mechanism that involves 1) direct recruitment and translational repression of genes that promote spermatogonial differentiation, and 2) repression of the target of rapamycin complex 1 (mTORC1), a well-known negative pathway for SSC self-renewal, by sequestration of the core factor mTOR in mRNPs. This mechanism links mRNA turnover to mTORC1 signaling through Nanos2-containing mRNPs and establishes a post-transcriptional buffering system to facilitate SSC homeostasis in the fluctuating environment within the seminiferous tubule. This research is partly supported by Grant-in-Aid for Scientific Research on Innovative Areas ”Epigenome dynamics and regulation in germ cells” and a “Data assimilation” project of the Transdisciplinary Research Integration Center in ROIS. Zhi Zhou was a NIG postdoctoral fellow 2012.
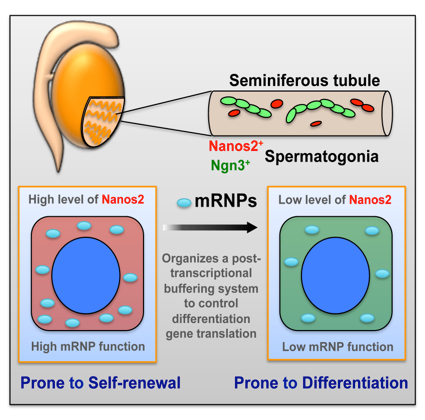
Nanos2 promotes mRNP formation and contribute to the maintenance of undifferentiated state of spermatogonial stem cells. Upon decreasing of Nanos2 level, stem cells shift to differentiation pathway.
Division of Brain Function / Hirata Group
Chemokine signalling controls integrity of radial glial scaffold in developing spinal cord and consequential proper position of boundary cap cells
Yan Zhu, Tomoko Matsumoto, Takashi Nagasawa, Fabienne Mackay, Fujio Murakami Journal of Neuroscience 17 June 2015, 35(24): 9211-9224; DOI:10.1523/JNEUROSCI.0156-15.2015Radial glial cells, the CNS neural progenitors, extend long radial processes that form the radial glial scaffold in the developing neuroepithelium. The integrity of this radial glial scaffold is well maintained throughout development. However, how this is achieved while the neuroepithelium rapidly expands and what the consequence might be when this integrity is compromised have not been well understood. In this study, we addressed these questions in the developing mouse spinal cord. We found that CXCR4, a receptor of a chemokine CXCL12 secreted from the pial meninges, is expressed in spinal cord radial glia. Conditional knockout of CXCR4 in radial glia causes disrupted radial glial scaffold with gaps at the pial endfeet layer, and consequentially lead to an invasion of boundary cap cells into the spinal cord. Since boundary cap cells are PNS cells normally positioned at the incoming and outgoing axonal roots, their invasion into the spinal cord suggests a compromised CNS/PNS boundary in the absence of CXCL12/CXCR4 signalling. By interrogating the underlying mechanisms, we then went on to show that CXCL12 signalling promotes the radial glia adhesion to basement membrane components and activates integrin β1 avidity. Our study uncovers an extrinsic molecular regulator for the maintenance of radial glial scaffold integrity during development. And our data suggest that the integrity of radial glia scaffold is important to safeguard the CNS/PNS boundary for the developing spinal cord.
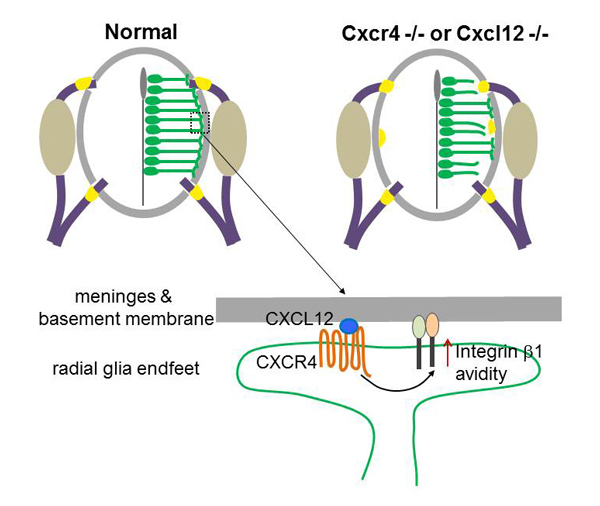
CXCL12 from the meninges regulates adhesion of radial glia to the basement membrane by enhancing avidity of integrin β1. Mice deficient in CXCL12 or its receptor CXCR4 show disrupted radial glial scaffold in their spinal cords, which consequentially causes invasion of boundary cap cells into the spinal cord.
NIG_MoG (The National Institute of Genetics Mouse Genome database)
URL: http://molossinus.lab.nig.ac.jp/msmdb/
Mammalian Genetics Laboratory / Shiroishi Group
The National Institute of Genetics Mouse Genome database (NIG_MoG; http://molossinus.lab.nig.ac.jp/msmdb/) primarily comprises the whole genome sequence data of two inbred mouse strains, MSM/Ms and JF1/Ms. These strains were established at NIG and originated from the Japanese subspecies Mus musculus molossinus. A comparative analysis of their genomes with C57BL/6J (B6: whose genome is predominantly derived from the West European subspecies M. m. domesticus), revealed a vast number of SNPs, structural variants, insertions and deletions (indels). NIG_MoG provides, especially for wet-lab biologists, visualized genome polymorphism information, browsing single-nucleotide polymorphisms and short insertions and deletions in the genomes of MSM/Ms and JF1/Ms allowing users to intuitively recognize intersubspecific genome divergence in these mouse strains using visual data. The database also supports the in silico screening of BAC clones that contain genomic DNA from MSM/Ms and the standard classical laboratory strain C57BL/6N established at RIKEN BioResorce center. NIG_MoG is thus a valuable navigator for exploring mouse genome polymorphisms and BAC clones that are useful for studies of gene function and regulation based on intersubspecific genome divergence.
Fig. A screenshot of the top page of NIG_MoG
Biological Macromolecules Laboratory • Maeshima Group
Chromosomes Progress to Metaphase in Multiple Discrete Steps via Global Compaction/Expansion Cycles
Liang, Z., Zickler, D., Prentiss, M., Chang. F.S., Witz, G., Maeshima, K., Kleckner, N. Cell, 161, 1124-1137 (2015). DOI:10.1016/j.cell.2015.04.030Mammalian mitotic chromosome morphogenesis was analyzed by 4D live-cell and snapshot deconvolution fluorescence imaging. Prophase chromosomes, whose organization was previously unknown, are revealed to comprise co-oriented sister linear loop arrays displayed along a single, peripheral, regularly kinked topoisomeraseII /cohesin /condensinII axis (Fig a). Thereafter, rather than smooth, progressive compaction as generally envisioned, progression to metaphase is a discontinuous process involving chromosome expansion as well as compaction. At late prophase (Fig b), dependent on topoisomerase II and with concomitant cohesin release, chromosomes expand, axes split and straighten, and chromatin loops transit to a radial disposition around now-central axes. Finally, chromosomes globally compact, giving the metaphase state (Fig c). These patterns are consistent with the hypothesis that the molecular events of chromosome morphogenesis are governed by accumulation and release of chromosome stress, created by chromatin compaction and expansion. Chromosome state could evolve analogously throughout the cell cycle.
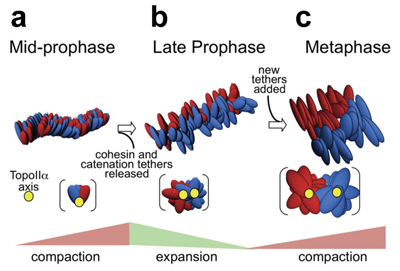
Two sister DNAs are shown in red and blue. The chromosome axis is in yellow. (a) mid-prophase; two sister chromatids are condensed with a single axis (yellow), but still mixed. (b) late-prophase; with expansion, two sisters and their axes are segregated. (c) metaphase; Further condensation and separation of the two sisters occur in metaphase.
Time & Date
9:00 ~ 16:00, Saturday, April 4th (No Reservations Required, Free Admission)
Access
Free Shuttle Buses Available to the National Institute of Genetics from the North Exit of Mishima Station. ( Service Time: 8:50 ~ 15:00)
Map
Map Free Shuttle Buses Available Parking around Mishima Station Available for Car Visitors.
No Pets Allowed except Service Dogs, No General Parking*
(*Disabled Parking Available on the Institute Premises)
Information
1111 Yata, Mishima, Shizuoka 411-8540, JAPAN TEL:+81-55-981-5873
Division of Agricultural Genetics • Kakutani Group
Genome-wide negative feedback drives transgenerational DNA methylation dynamics in Arabidopsis.
Tasuku Ito, Yoshiaki Tarutani, Taiko Kim To, Mohamed Kassam, Evelyne Duvernois-Berthet, Sandra Cortijo, Kazuya Takashima, Hidetoshi Saze, Atsushi Toyoda, Asao Fujiyama, Vincent Colot, Tetsuji Kakutani PLoS Genetics Published: April 22, 2015 DOI:10.1371/journal.pgen.1005154DNA methylation is important for controlling activity of transposable elements and genes. An intriguing feature of DNA methylation in plants is that its pattern can be inherited over multiple generations at high fidelity in a Mendelian manner. However, mechanisms controlling the trans-generational DNA methylation dynamics are largely unknown. Arabidopsis mutants of a chromatin remodeler gene DDM1 (Decrease in DNA Methylation 1) show drastic reduction of DNA methylation in transposons and repeats, and also show progressive changes in developmental phenotypes during propagation through self-pollination. We now show using whole genome DNA methylation sequencing that upon repeated selfing, the ddm1 mutation induces an ectopic accumulation of DNA methylation at hundreds of loci. Remarkably, even in the wild type background, the analogous de novo increase of DNA methylation can be induced in trans by chromosomes with reduced DNA methylation. Collectively, our findings support a model to explain the transgenerational DNA methylation redistribution by genome-wide negative feedback, which should be important for balanced differentiation of DNA methylation states within the genome. This work is a collaboration with Fujiyama lab supported by Systems Functional Genetics Project of the Transdisciplinary Research Integration Center, ROIS.
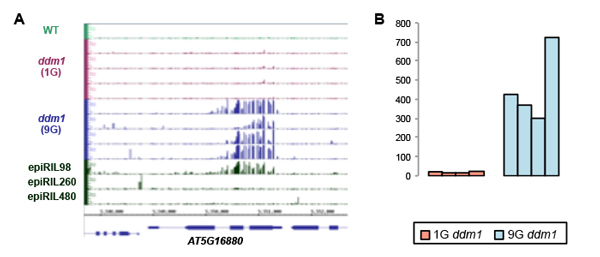
(A) Ectopic increase of cytosine methylation in the ddm1 mutant plants. The methylation accumulated in the 9th generation of ddm1 mutant (9G), but not in the 1st generation (1G). A line with large amount of chromosomes from ddm1 (epiRIL98) introduced into DDM1 wild type background also showed the ectopic DNA methylation. (B) Similar ectopic DNA methylation can be seen at hundreds of loci specifically in the 9G ddm1 mutant plants. The value shows number of genes with ectopic methylation in each of four 9G and four 1G ddm1 lines.
Division of Evolutionary Genetics • Akashi Group
Division of Population Genetics • Saitou Group
Whole-genome sequencing of six Mauritian cynomolgus macaques (Macaca fascicularis) reveals a genome-wide pattern of polymorphisms under extreme population bottleneck
Naoki Osada, Nilmini Hettiarachchi, Isaac Adeyemi Babarinde, Naruya Saitou, Antoine Blancher
Genome Biology and Evolution, 7(3):821–830 (2015) DOI: 10.1093/gbe/evv033
Cynomolgus macaques (Macaca fascicularis) are important animals for biomedical research and known to be genetically highly heterogeneous. They are mainly distributed throughout Southeast Asia, but a small number of individuals were introduced to the island of Mauritius by humans around the 16th century. The unique demographic history of the Mauritian cynomolgus macaques would be useful for understanding the effect of an extreme population bottleneck on the pattern of polymorphisms in genomes. We sequenced and analyzed the whole genomes of six Mauritian cynomolgus macaques, and found the mild reduction of overall level of nucleotide diversity as well as the strong reduction of low-frequency polymorphisms in the populations. In addition, we confirmed that the Mauritian cynomolgus macaques have been originated from Indonesia-Malaysian cynomolgus macaques. The information of the genome sequences would be useful for future biomedical research using those macaques that have unique population history.

(Left panel) The island of Mauritius is marked by the red circle; (Right panel) a picture of cynomolgus macaque
Biological Macromolecules Laboratory • Maeshima Group
End-targeting proteomics of isolated chromatin segments of a mammalian ribosomal RNA gene promoter
Ide, S. and Dejardin, J.he unbiased identification of proteins associated with specific loci is crucial for understanding chromatin-based processes. The proteomics of isolated chromatin fragment (PICh) method has previously been developed to purify telomeres and identify associated proteins. This approach is based on the affinity capture of endogenous chromatin segments by hybridization with oligonucleotide containing locked nucleic acids. However, PICh is only efficient with highly abundant genomic targets, limiting its applicability. Here we develop an approach for identifying factors bound to the promoter region of the ribosomal RNA genes that we call end-targeting PICh (ePICh). Using ePICh, we could specifically enrich the RNA polymerase I pre-initiation complex, including the selectivity factor 1. The high purity of the ePICh material allowed the identification of ZFP106, a novel factor regulating transcription initiation by targeting RNA polymerase I to the promoter. Our results demonstrate that ePICh can uncover novel proteins controlling endogenous regulatory elements in mammals.
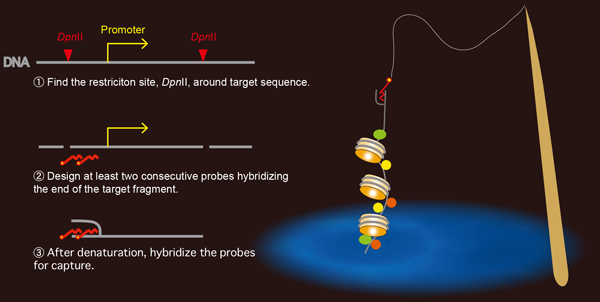
Rules for designing capture probes for the formation of stable oligo-DNA hybrids.
Genome Biology Laboratory • Kohara Group
Co-expression of the transcription factors CEH-14 and TTX-1 regulates AFD neuron-specific genes gcy-8 and gcy-18 in C. elegans.
Hiroshi KAGOSHIMA and Yuji KOHARAA wide variety of cells are generated by the expression of characteristic sets of genes, primarily those regulated by cell-specific transcription. To elucidate the mechanism regulating cell-specific gene expression in a highly specialized cell, AFD thermosensory neuron in Caenorhabditis elegans, we analyzed the promoter sequences of guanylyl cyclase genes, gcy-8 and gcy-18, exclusively expressed in AFD. In this study, we showed that AFD-specific expression of gcy-8 and gcy-18 requires the co-expression of homeodomain proteins, CEH-14/LHX3 and TTX-1/OTX1. We observed that mutation of ttx-1 or ceh-14 caused a reduction in the expression of gcy-8 and gcy-18 and that the expression was completely lost in double mutants. This synergy effect was also observed with other AFD marker genes, such as ntc-1, nlp-21, and cng-3. Electrophoretic mobility shift assays revealed direct interaction of CEH-14 and TTX-1 proteins with gcy-8 and gcy-18 promoters in vitro. The binding sites of CEH-14 and TTX-1 proteins were confirmed to be essential for AFD-specific expression of gcy-8 and gcy-18 in vivo. We also demonstrated that forced expression of CEH-14 and TTX-1 in AWB chemosensory neurons induced ectopic expression of gcy-8 and gcy-18 reporters in this neuron. Finally, we showed that the regulation of gcy-8 and gcy-18 expression by ceh-14 and ttx-1 is evolutionally conserved in five Caenorhabditis species. Taken together, ceh-14 and ttx-1 expression determines the fate of AFD as terminal selector genes at the final step of cell specification.
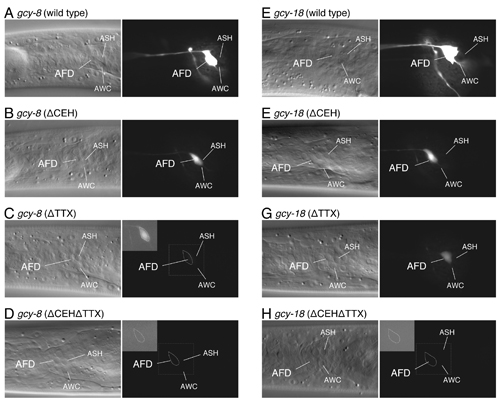
Expression analysis of gcy-8 and gcy-18 promoter GFP constructs with CEH-14 and/or TTX-1 binding site mutations.
GFP reporter expression of gcy-8 (A-D) and gcy-18 (E-H) promoter sequences with CEH-14 and/or TTX-1 binding site mutations; (wild-type: A, E) wild-type 300-bp promoter, (ΔCEH: B, F) CEH-14 binding site mutation, (ΔTTX: C, G) TTX-1 binding site mutation, and (ΔCEHΔTTX: D, H) CEH-14 and TTX-1 binding site mutation. Images were acquired using an exposure time of 0.2 s (A, E) and 1 s (B-D, F-H). The inset shows digitally enhanced images of the AFD cell body (shown by the dotted line).
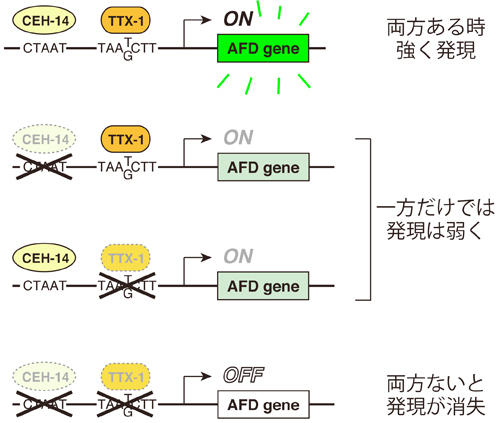
CEH-14 and TTX-1 binding sites are essential for gcy-8 and gcy-18 expression in AFD
CEH-14 and TTX-1 directly bind to promoters of the AFD genes (CEH-14: CTAAT, TTX-1: TAA(T/G)CTT) and co-regulate downstream genes. The expression was reduced in GFP reporters carrying either of CEH-14 or TTX-1 binding site mutation, whereas reporter carrying double mutation (ΔCEHΔTTX) completely lost GFP expression.
Motor Neural Circuit Laboratory • Hirata Group
RING finger protein 121 facilitates the degradation and membrane localization of voltage-gated sodium channels
Ogino, K., Low, S. E., Yamada, K., Saint-Amant, L., Zhou, W., Muto, A., Asakawa, K., Nakai, J., Kawakami, K., Kuwada, J. Y., and Hirata, H.Voltage-gated sodium channels (NaV) are known to form clusters at the membranes of excitable cells; however, what governs their transport is largely unknown. We found that the endoplasmic reticulum (ER) and cis-Golgi associated ubiquitin ligase really interesting new gene (RING) finger protein 121 (RNF121) mediates the degradation and membrane localization of NaV. This apparent quality control of NaV ensures the transport of properly folded channels to the membranes of excitable cells. To our knowledge, this is the first pathologically relevant identification of a voltage-gated ion channel as a substrate for ER-associated protein degradation, whose degradation is governed by an ER- and Golgi-associated E3-ubiquitin ligase.
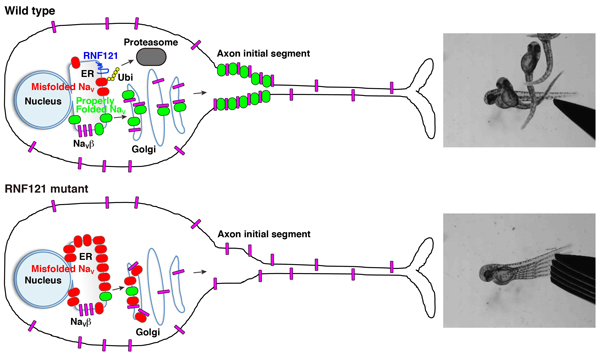
A wild-type neuron wherein RNF121 mediates ubiquitination of misfolded NaV channels marking them for proteasome-mediated degradation. Properly folded NaV channels (green) associate with NaVβ subunits (magenta) in the Golgi apparatus and are transported to the axon initial segment. Of note, some NaVβ subunits are transported to the membrane independent of NaV channels. An RNF121-deficient neuron wherein misfolded NaV channels (red) accumulates in the ER and cis-Golgi compartments, which, over time, depletes NaVβ subunits, preventing them from forming complexes with properly folded NaV channels, causing an impairment of NaV transport.
Power law relationship between cell cycle duration and cell volume in the early embryonic development of Caenorhabditis elegans
Yukinobu Arata, Hiroaki Takagi, Yasushi Sako, and Hitoshi SawaCell cycle is regulated in coordination with cell size. Yoshio Masui at Toronto University reported that the cell cycle duration is inversely proportional to the cell radius squared after the MBT in Xenopus embryo. They proposed that cell surface area is a candidate to determine cell cycle duration. However, it remains unknown whether this “second power law” is conserved in other animal embryos. Here, we found that the relationship between cell cycle duration and cell size in Caenorhabditis elegans embryos exhibited a power law distribution. The powers of the time-size relationship could be grouped into at least three classes according to C. elegans founder cell lineages. Any of the values was less than that in Xenopus embryos. Interestingly, we found that the relationship between cell and nuclear volumes exhibited a power law. Supposing interactions between nuclease and cytoplasm, we considered the volume ratio between nucleus and cell. The value of power in the volume ratio was almost identical to that of the time-size relationship in highly-size correlated lineages. We propose a new model that cell cycle duration in C. elegans embryos is determined by a geometric constraint between intercellular structures such as nucleus and cytoplasm. It remains possible that cell cycle durations in other animal embryos are determined by the same constraint.
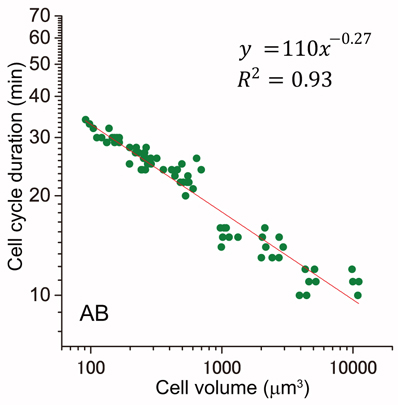
Cell cycle duration and cell volume in the AB lineage exhibited a linear relationship in a double logarithmic plot (power law).
Biological Macromolecules Laboratory • Maeshima Group
(i) Structural Evaluation of Tandem Hairpin Pyrrole–Imidazole Polyam-ides Recognizing Human Telomeres
Hirata, A., Nokihara, K., Kawamoto, Y., Bando, T., Sasaki, A., Ide, S., Maeshima, K., Kasama, T., and Sugiyama, H. Journal of the American Chemical Society (JACS), July 18, 2014. DOI: 10.1021/ja506058e
(ii) Tandem Trimer Pyrrole-Imidazole Polyamide Probes Targeting 18 Base Pairs in Human Telomere Sequences
Kawamoto, Y., Sasaki, A., Hashiya, K., Ide, S., Bando, T. *, Maeshima, K. *, and Sugiyama, H.*Pyrrole-Imidazole (PI) polyamides bind to the minor groove of DNA in a sequence-specific manner. Our previous studies have demonstrated that a synthesized tandem hairpin PI polyamide (TH59) can target human telomere sequences (TTAGGG)n under mild conditions and can serve as a new probe for studying human telomeres. In the recently published two papers, we have improved specificity of the tandem hairpin PI polyamide by optimization of the connecting region (hinge region) of the tandem hairpin parts [HPTH59-b in Fig. A and paper (i)] and by introducing additional hairpin part [TT59 in Fig. B and paper (ii)]. The HPTH59-b and TT59 have higher specificity to recognize human telomeric repeats than the previous ones and stain telomeres in chemically fixed human cells with lower background signal.
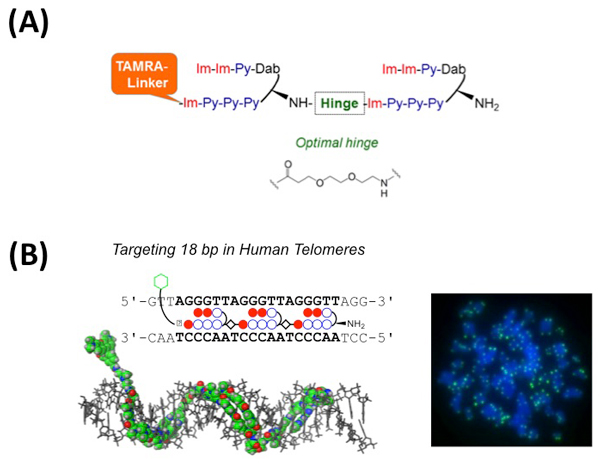
(A) Schematic structure of HPTH59-b. The optimized chemical structure of hinge segment is shown.
(B) Left, schematic representation of the tandem trimer PI polyamide TT59, which binds to 18bp of the human telomere sequence. Right, the telomere regions of chromosome ends are labeled with TT59 (green). DNA stain (blue).
The Royal Swedish Academy of Sciences has awarded the 2015 Crafoord Prize in Biosciences to Professor emeritus Tomoko Ohta and Professor emeritus Richard Lewontin for their pioneering work in understanding genetic variation and evolution. The award ceremony will take place on May 5-7 in Stockholm, Sweden. The Crafoord Prize in astronomy and mathematics, biosciences, geosciences or polyarthritis research is awarded annually according to a rotating scheme. These disciplines are chosen to complement those for which the Nobel Prizes are awarded.
This year’s prize recognizes Professor Ohta’s fundamental contributions to the understanding of genetic polymorphism conducted at NIG. Her work, including “Slightly Deleterious Mutant Substitutions in Evolution” published by Nature in 1973 proposed “near neutrality” as a important factor for understanding both variation within a population and differences among species. Weak selection is now considered central to understanding genome evolution and her ideas have permeated thinking in biomedical research and systems biology as well as in comparative genomics.
Biological Macromolecules Laboratory • Maeshima Group
The physical size of transcription factors is key to transcriptional regulation in chromatin domains
Kazuhiro Maeshima, Kazunari Kaizu, Sachiko Tamura, Tadasu Nozaki, Tetsuro Kokubo, and Koichi TakahashiGenetic information, which is stored in the long strand of genomic DNA as chromatin, must be scanned and read out by various transcription factors. First, gene-specific transcription factors, which are relatively small (~50 kDa), scan the genome and bind regulatory elements. Such factors then recruit general transcription factors, Mediators, RNA polymerases, nucleosome remodellers, and histone modifiers, most of which are large protein complexes of 1–3 MDa in size. Here, we propose a new model for the functional significance of the size of transcription factors (or complexes) for gene regulation of chromatin domains. Recent findings suggest that chromatin consists of irregularly folded nucleosome fibres (10 nm fibres) and forms numerous condensed domains (e.g., topologically associating domains)(blue balls in Fig). Although the flexibility and dynamics of chromatin allow repositioning of genes within the condensed domains, the size exclusion effect of the domain may limit accessibility of DNA sequences by transcription factors. We used Monte Carlo computer simulation to determine the physical size limit of transcription factors that can enter condensed chromatin domains. Small gene-specific transcription factors can penetrate into the chromatin domains and search their target sequences, whereas large transcription complexes cannot enter the domain (Fig a). Due to this property, once a large complex binds its target site via gene-specific factors it can act as a “buoy” to keep the target region on the surface of the condensed domain (Fig b) and maintain transcriptional competency (Fig c). This size-dependent specialisation of target-scanning and surface-tethering functions could provide novel insight into the mechanisms of various DNA transactions, such as DNA replication and repair/recombination.
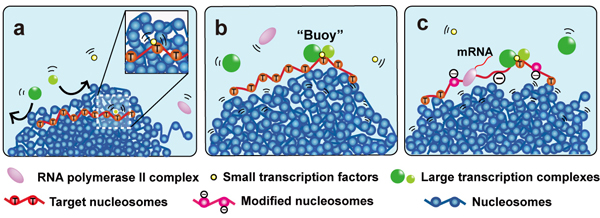
“Buoy” Model for Transcriptional Regulation
Condensed chromatin domains are shown with blue nucleosomes.
(a) The small transcription factors in yellow can move in the condensed chromatin domain but not large transcription complexes (in green). (b) The small transcription factor bound to the target region (red nucleosomes) and can recruit large transcription complexes when it is relocated on the domain surface. Binding of large transcription complexes (green) keeps the transcriptional region (red nucleosomes) on the domain surface like a ‘buoy’. (c) With other large complexes such as nucleosome remodeler or histone modifier, stable transcription is maintained.
超分子構造研究室・白木原研究室
Structural basis for replication origin unwinding by an initiator-primase of plasmid ColE2-P9: Duplex DNA unwinding by a single protein
Hiroshi Itou, Masaru Yagura, Yasuo Shirakihara, and Tateo ItohDuplex DNA is generally unwound by protein oligomers prior to replication. The Rep protein of plasmid ColE2-P9 is an essential initiator for plasmid DNA replication. This protein binds the replication origin (Ori) in a sequence specific manner as a monomer and unwinds DNA. We present the crystal structure of the DNA-binding domain of Rep (E2Rep-DBD) in complex with Ori DNA. The structure unveils the basis for Ori specific recognition by the E2Rep-DBD and also reveals that it unwinds DNA by the concerted actions of its three contiguous structural modules. The structure also shows that the functionally unknown PriCT domain, which forms a compact module, plays a central role in DNA unwinding. The conservation of the PriCT domain in the C-termini of some archaeo-eukaryotic primases, indicates that it likely plays a similar role in these proteins. Thus, this is the first report providing the structural basis for the functional importance of the conserved PriCT domain and also reveals a novel mechanism for DNA unwinding by a single protein.
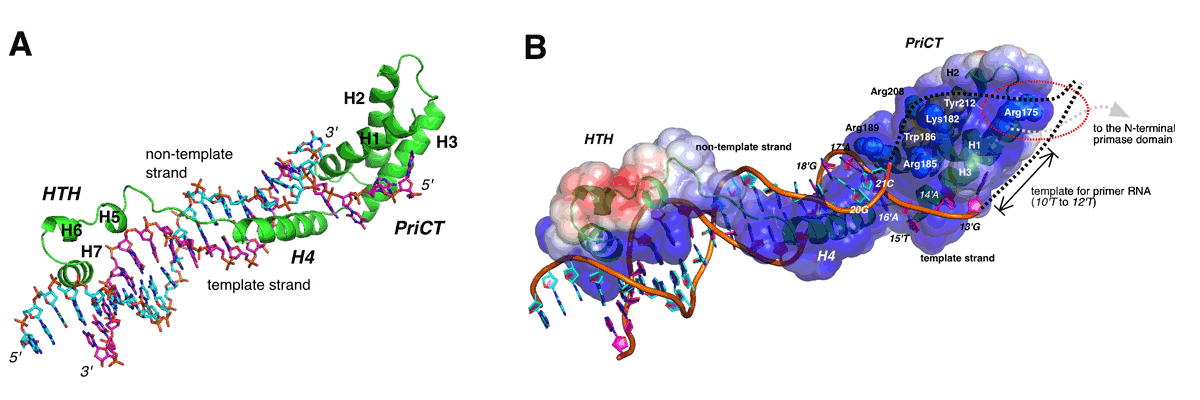
(A) The ribbon model of the complex consists of E2Rep-DBD and Ori DNA. Elongated fold of E2Rep-DBD enables binding specificity and affinity enough to unwind duplex DNA.
(B) Model for unwinding of the complete Ori by Rep. The PriCT-module plays a central role in unwinding of duplex DNA and stabilizing the single-stranded DNA.










