



Sakai Group / Model Fish Genetics Laboratory
Biallelic TEDC1 variants cause a new syndrome with severe growth impairment and endocrine complications
Noriko Miyake, Kentaro Shiga, Yuya Hasegawa, Chisato Iwabuchi, Kohei Shiroshita, Hiroshi Kobayashi, Keiyo Takubo, Fabien Velilla, Akiteru Maeno, Toshihiro Kawasaki, Yukiko Imai, Noriyoshi Sakai, Tomonori Hirose, Atsushi Fujita, Hidehisa Takahashi, Nobuhiko Okamoto, Mikako Enokizono, Shiho Iwasaki, Syuichi Ito, Naomichi Matsumoto
European Journal of Human Genetics 2025 Feb 20. DOI:10.1038/s41431-025-01802-3
We encountered two affected male patients born to non-consanguineous parents, who presented with prenatal-onset severe growth impairment, primary microcephaly, developmental delay, adrenal insufficiency, congenital glaucoma, delayed bone aging, craniosynostosis, congenital tracheal stenosis, and primary hypogonadism. By exome sequencing, we identified compound heterozygous TEDC1 variants (NM_001134877.1 c.[104-5C>G];[787delG] p.[?];[(Ala263LeufsTer29)] in both affected siblings. We confirmed that the splice site variant, c.104-5C>G leads to no TEDC1 protein production via nonsense-mediated mRNA decay. The frameshift variant located in the last coding exon, c.787delG, produces a C-terminally truncated protein, which impairs the binding with TEDC2. Thus, both variants are thought to be loss-of-function. TEDC1 and TEDC2 are both required for centriole stability and cell proliferation. Our in vitro experiments using patient-derived cells revealed cell cycle abnormality. Our in vivo study using tedc1−/− zebrafish generated by CRISPR/Cas9 successfully recapitulated the growth impairment and cranial bone dysplasia as seen in our patients. The tedc1−/− mutant zebrafish were sterile and did not have developed gonads. Furthermore, we showed that biallelic TEDC1 deletion causes cilia abnormalities through defective acetylated tubulins.
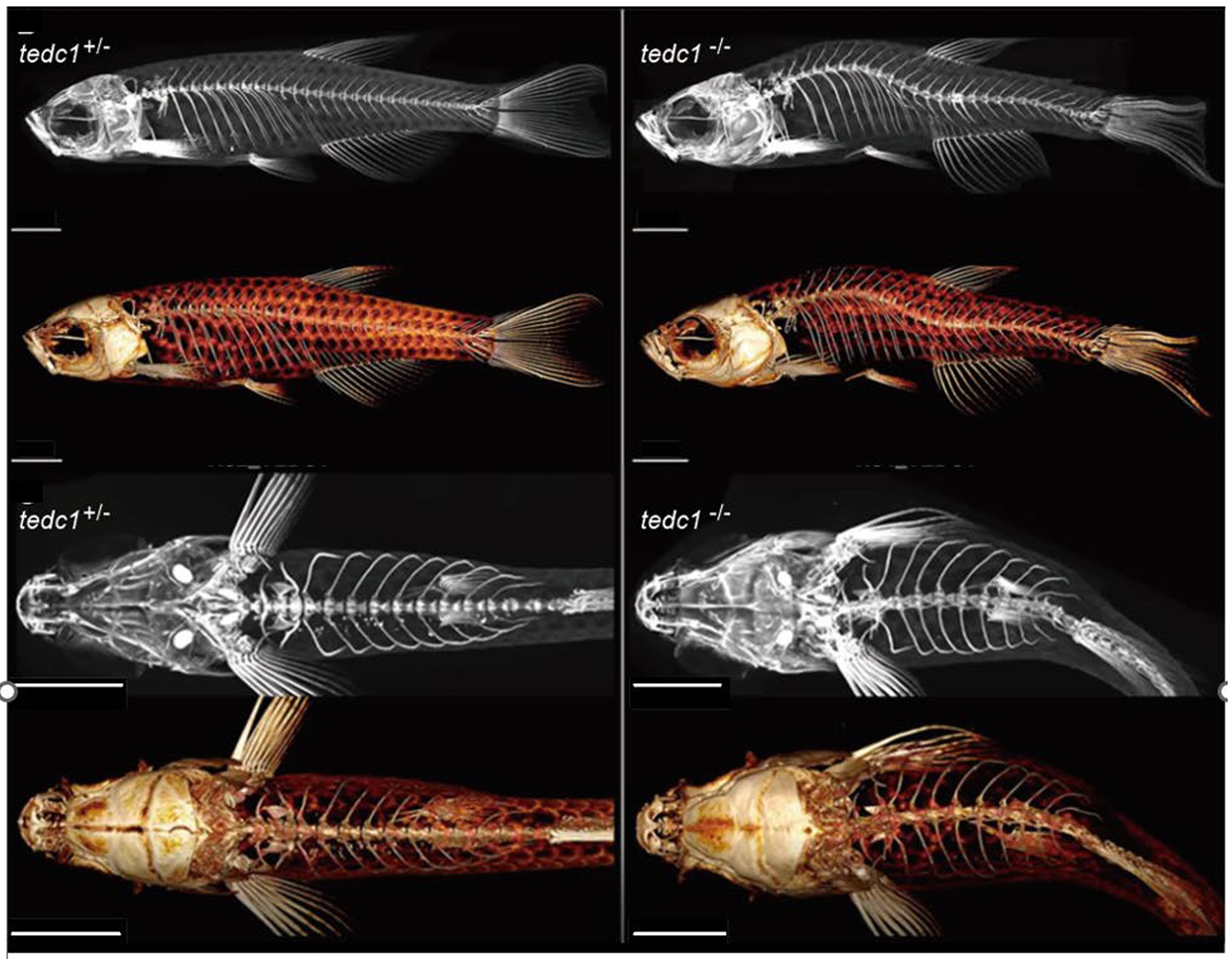
Figure: Representative lateral and upper view µCT images of tedc1+/− and tedc1−/− zebrafish
Maeshima Group / Genome Dynamics Laboratory
Chromatin domains in the cell: phase separation and condensation
Shin Fujishiro*, Masaki Sasai*, and Kazuhiro Maeshima*
*Corresponding author
Current Opinion in Structural Biology Volume 91, April 2025, 103006 DOI:10.1016/j.sbi.2025.103006
Free download link for 50 days [Available online 20 February 2025]
Negatively charged genomic DNA wraps around positively charged core histone octamers to form nucleosomes, which, along with proteins and RNAs, self-organize into chromatin within the nucleus. In eukaryotic cells, chromatin forms loops that collapse into chromatin domains and serve as functional units of the genome. Since various factors—such as chromatin-binding proteins, histone modifications, transcriptional states, depletion attraction, and cations—can significantly impact chromatin organization, the formation processes of these hierarchical structures remain unclear. In this review paper, Shin Fujishiro (Kyoto Univ), Masaki Sasai (Kyoto Univ) and Kazuhiro Maeshima (NIG) critically discuss the formation mechanisms of the chromatin domain in the cell from a physical point of view, including phase separation and condensation.
This work was supported by the JSPS and MEXT KAKENHI grants (JP20H05936, JP22H00406, JP23K17398, and JP24H00061), and the Takeda Science Foundation.
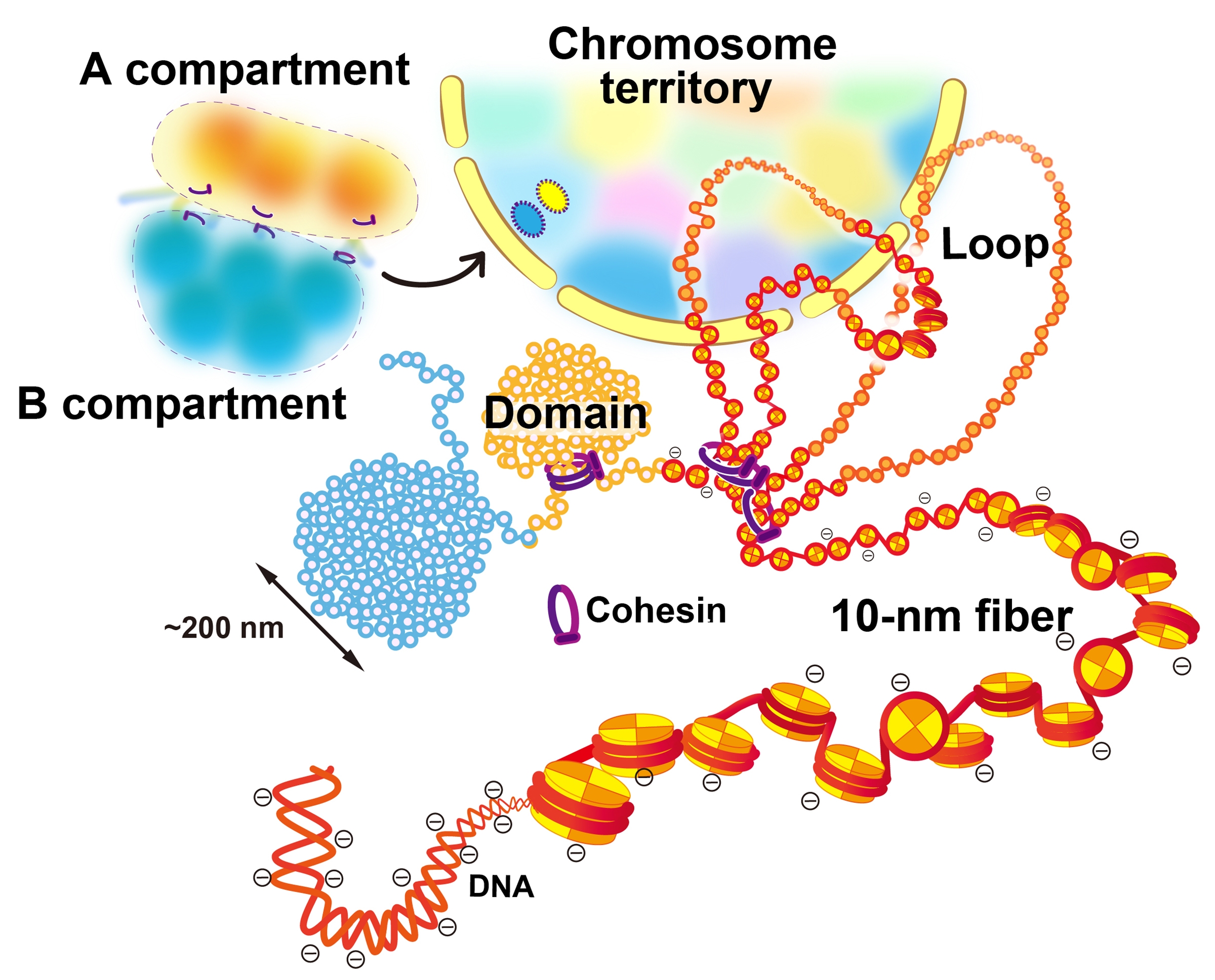
Figure: Scheme for hierarchical chromatin organization inside the cell nucleus.
DNA is wrapped around histone octamer to make a nucleosome. The chain of nucleosomes is compacted into chromatin domains, which interact over long distances to form chromatin compartments. A and B compartments in general represent transcriptionally active or open chromatin state (compartment A) and inactive or closed chromatin (compartment B), respectively. A single interphase chromosome consists of several compartments occupies that collectively form a chromosome territory. Note that this scheme is highly simplified and a more complex organization can be possible inside the cell.
Press release
Swirling Instability mediated by Elastic and Hydrodynamic Couplings in Cytoplasmic Streaming
Takuji Ishikawa*, Takayuki Torisawa, Hirofumi Wada, Akatsuki Kimura*.
*corresponding authors
PRX Life (2025) 3, 013008 DOI:10.1103/PRXLife.3.013008
![]() Press release (In Japanese only)
Press release (In Japanese only)
Cells contain numerous molecules that collectively function to maintain cellular activity. One striking example is cytoplasmic streaming, which is coordinated intracellular flow. Although individual molecules interact locally, how their movements collectively form an organized flow remains a mystery. In 2017, Prof. Kimura’s team studied cytoplasmic streaming in C. elegans zygotes and discovered that microtubules align via endoplasmic reticulum (ER) connections, and direct flow. At that time, Prof. Joanny (Curie Institute) developed a 1D mathematical model to explain flow initiation and reversal. However, a 3D simulation is required for more detailed comparison with real cells.
In the present study, Prof. Ishikawa (Tohoku Univ.) created a 3D computational model, refined through collaboration with Prof. Wada (Ritsumeikan Univ.). This revealed that ER elasticity is crucial—too weak, flow ceases, too strong, reversals disappear. These results matched the experimental data obtained by Dr. Torisawa (Cell Architecture Lab, NIG). This study quantitatively explains cytoplasmic streaming dynamics and provides a foundation for understanding intracellular flow in various organisms.
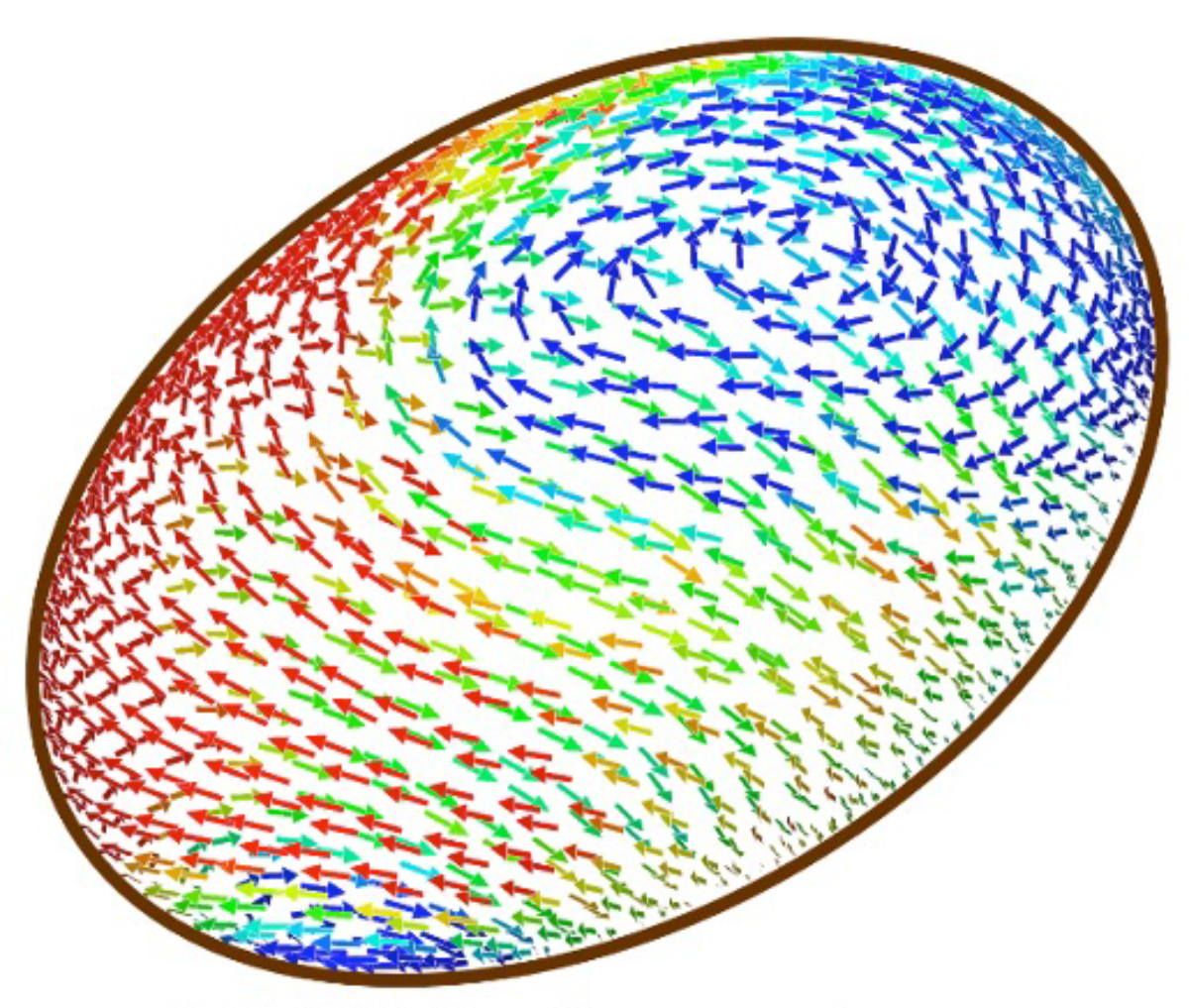
Figure: A snapshot of the simulation constructed in this study. The ellipsoid represents the fertilized egg of the nematode. The direction and speed of the flow near the surface of the cell are indicated by the direction and the colors of the arrows (red=fast, blue=slow). The image adopted from Fig. 2b of the paper.
Kitano Group / Ecological Genetics Laboratory
River dam prevents the invasion of non-native species of Neocaridina Kubo, 1938 (Decapoda: Caridea: Atyidae) into native habitats: A case study in the Yumesaki River system, Japan
Ryosuke Ishii and Yusuke Fuke *
* Corresponding author
Journal of Crustacean Biology (2025) 45, ruaf009 DOI:10.1093/jcbiol/ruaf009
The range expansion of invasive species that threaten biodiversity is caused by repeated cycles of their artificial introduction and subsequent dispersal. Dispersal can occur without human intervention, and its pattern is mainly determined by environmental factors and species characteristics.
In this study, Yusuke Fuke, JSPS postdoctoral fellow at the National Institute of Genetics, and Ryosuke Ishii, master’s student at Graduate School of Agriculture, Kagawa University, focused on the freshwater shrimp community in the Yumesaki River system, Japan, which is the habitat of the native species Neocaridina denticulata and has experienced the introduction of the closely related non-native species N. davidi. They examined the genetic population structures and morphological characteristics of these species to elucidate their dispersal process. Their results showed that populations comprising only the native species remained upstream of the dam, whereas the native species was replaced by the non-native species at the other sites. These results demonstrate that the distribution of non-native species can expand throughout the entire river system in rivers without structures that restrict the movement of aquatic organisms.

Figure: Native freshwater shrimp Neocaridina denticulata, found only upstream of the dam in the water system studied in this study.
Press release
A Near Complete Genome Assembly of the Oshima Cherry Cerasus speciosa
Kazumichi Fujiwara, Atsushi Toyoda, Bhim B. Biswa, Takushi Kishida, Momi Tsuruta, Yasukazu Nakamura, Noriko Kimura, Shoko Kawamoto, Yutaka Sato, Toshio Katsuki, Sakura 100 Genome Consortium, and Tsuyoshi Koide*
Sakura 100 Genome Consortium (full list): Kazumichi Fujiwara, Atsushi Toyoda, Bhim B. Biswa, Takushi Kishida, Momi Tsuruta, Yasukazu Nakamura, Noriko Kimura, Shoko Kawamoto, Yutaka Sato, Toshio Katsuki, Tsuyoshi Koide*, Akatsuki Kimura, Ken-Ichi Nonomura, Hironori Niki, Hiroyuki Yano, Kinji Umehara, Tazro Ohta, Chikahiko Suzuki.
*Corresponding Author
Scientific Data volume 12, Article number: 162 (2025) DOI:10.1038/s41597-025-04388-z
![]() Press release (In Japanese only)
Press release (In Japanese only)
The Oshima cherry (Cerasus speciosa), which is endemic to Japan, has significant cultural and horticultural value. In this study, we present a near complete telomere-to-telomere genome assembly for C. speciosa, derived from the old growth “Sakurakkabu” tree on Izu Oshima Island. Using Illumina short-read, PacBio long-read, and Hi-C sequencing, we constructed a 269.3 Mbp genome assembly with a contig N50 of 32.0 Mbp. We examined the distribution of repetitive sequences in the assembled genome and identified regions that appeared to be centromeric. Detailed structural analysis of these putative centromeric regions revealed that the centromeric regions of C. speciosa comprised repetitive sequences with monomer lengths of 166 or 167 bp. Comparative genomic analysis with Prunus sensu lato genome revealed structural variations and conserved syntenic regions. This high-quality reference genome provides a crucial tool for studying the genetic diversity and evolutionary history of Cerasus species, facilitating advancements in horticultural research and the preservation of this iconic species.










