



Kanemaki Group / Molecular Cell Engineering Laboratory
MCMBP promotes the assembly of the MCM2–7 hetero-hexamer to ensure robust DNA replication in human cells
Yuichiro Saito, Venny Santosa, Kei-ichiro Ishiguro and Masato T. Kanemaki.
eLife (2022) 11, e77393 DOI:10.7554/eLife.77393
Replication of the genetic material DNA is essential for cell proliferation. The MCM2–7 hexamer, a ring-like complex composed of six subunits from MCM2 to MCM7, functions as the replicative helicase for unwinding double-stranded DNA. It is known that a large amount of the MCM2–7 hexamer is required for efficient DNA replication in the S phase. However, how the MCM2–7 hexamer is assembled has not been understood.
This paper shows that the MCM-binding protein (MCMBP) binds to the MCM subunits and plays a crucial function in incorporating MCM3 and MCM5 into the hexamer (Figure 1). Rapid degradation of MCMBP using the auxin-inducible degron 2 system (AID2) resulted in a reduced expression of the functional MCM2–7 hexamer at each cell division because newly synthesized MCM3 was not incorporated into the hexamer.
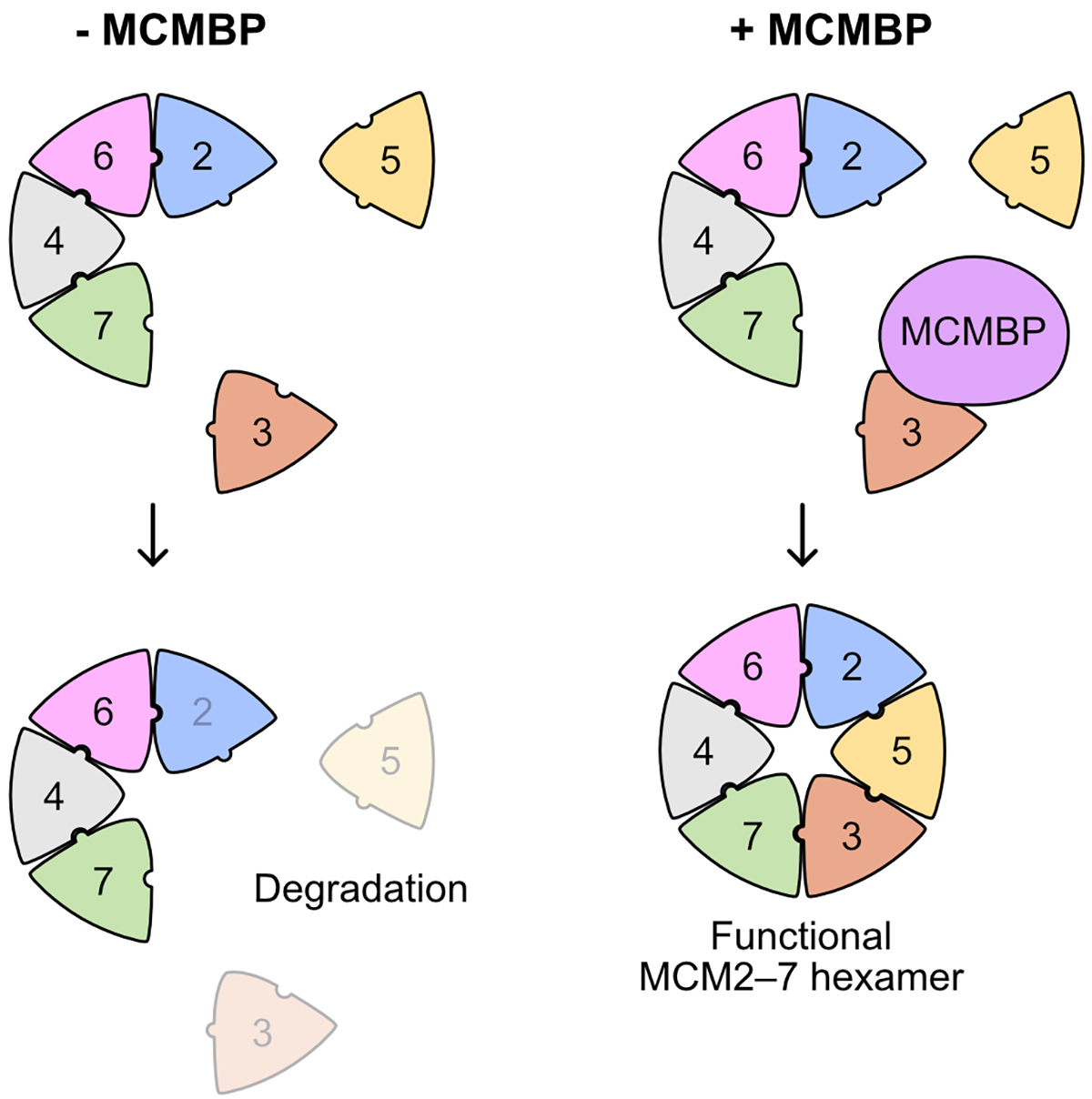
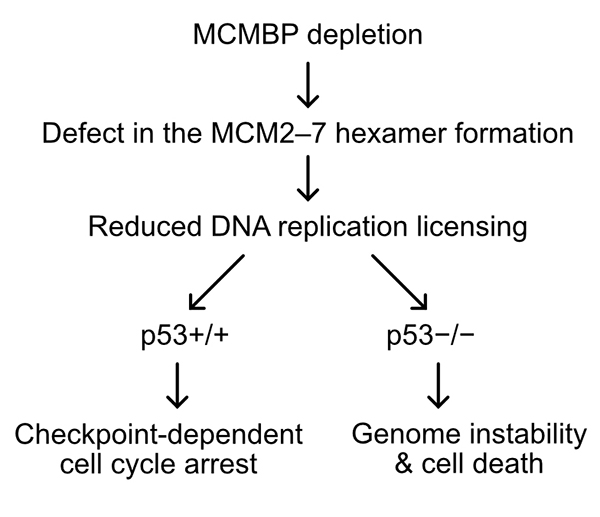
Interestingly, when the level of MCM2–7 hexamer decreased, human cells expressing the tumor suppressor gene p53 maintained genome integrity by transiently arresting the cell cycle in the G1 phase (Fig. 2). In contrast, cells lacking p53 induced cell death by entering the S phase with fewer hexamers resulting in incomplete DNA replication. These results suggest that depletion of MCMBP may specifically eliminate cancer cells with mutations in p53.
The Kanemaki Laboratory at National Institute of Genetics led this research in collaboration with Prof. Kei-ichiro Ishiguro at Kumamoto University.
Press release

Metagenomics reveals global-scale contrasts in nitrogen cycling and cyanobacterial light harvesting mechanisms in glacier cryoconite
T. Murakami, N. Takeuchi, H. Mori, Y. Hirose, A. Edwards, T. Irvine-Fynn, Z. Li, S. Ishii, T. Segawa
Microbiome (2022) 10, 50 DOI:10.1186/s40168-022-01238-7
![]() Press release (In Japanese only)
Press release (In Japanese only)
Background
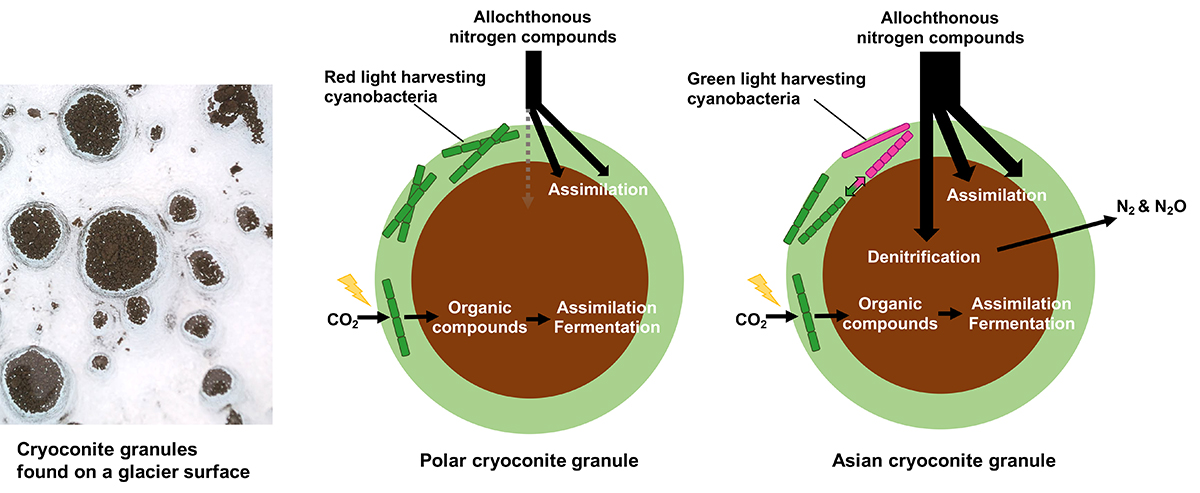
Cryoconite granules are mineral–microbial aggregates found on glacier surfaces worldwide and are hotspots of biogeochemical reactions in glacier ecosystems. However, despite their importance within glacier ecosystems, the geographical diversity of taxonomic assemblages and metabolic potential of cryoconite communities around the globe remain unclear. In particular, the genomic content of cryoconite communities on Asia’s high mountain glaciers, which represent a substantial portion of Earth’s ice masses, has rarely been reported. Therefore, in this study, to elucidate the taxonomic and ecological diversities of cryoconite bacterial consortia on a global scale, we conducted shotgun metagenomic sequencing of cryoconite acquired from a range of geographical areas comprising Polar (Arctic and Antarctic) and Asian alpine regions.
Results
Our metagenomic data indicate that compositions of both bacterial taxa and functional genes are particularly distinctive for Asian cryoconite. Read abundance of the genes responsible for denitrification was significantly more abundant in Asian cryoconite than the Polar cryoconite, implying that denitrification is more enhanced in Asian glaciers. The taxonomic composition of Cyanobacteria, the key primary producers in cryoconite communities, also differs between the Polar and Asian samples. Analyses on the metagenome-assembled genomes and fluorescence emission spectra reveal that Asian cryoconite is dominated by multiple cyanobacterial lineages possessing phycoerythrin, a green light-harvesting component for photosynthesis. In contrast, Polar cryoconite is dominated by a single cyanobacterial species Phormidesmis priestleyi that does not possess phycoerythrin. These findings suggest that the assemblage of cryoconite bacterial communities respond to regional- or glacier-specific physicochemical conditions, such as the availability of nutrients (e.g., nitrate and dissolved organic carbon) and light (i.e., incident shortwave radiation).
Conclusions
Our genome-resolved metagenomics provides the first characterization of the taxonomic and metabolic diversities of cryoconite from contrasting geographical areas, highlighted by the distinct light-harvesting approaches of Cyanobacteria and nitrogen utilization between Polar and Asian cryoconite, and implies the existence of environmental controls on the assemblage of cryoconite communities. These findings deepen our understanding of the biodiversity and biogeochemical cycles of glacier ecosystems, which are susceptible to ongoing climate change and glacier decline, on a global scale.
Source: T. Murakami et al., Microbiome DOI:10.1186/s40168-022-01238-7
Multilateral benefit-sharing from digital sequence information will support both science and biodiversity conservation
Amber Hartman Scholz, Jens Freitag, Christopher H. C. Lyal, Rodrigo Sara, Martha Lucia Cepeda, Ibon Cancio, Scarlett Sett, Andrew Lee Hufton, Yemisrach Abebaw, Kailash Bansal, Halima Benbouza, Hamadi Iddi Boga, Sylvain Brisse, Michael W. Bruford, Hayley Clissold, Guy Cochrane, Jonathan A. Coddington, Anne-Caroline Deletoille, Felipe García-Cardona, Michelle Hamer, Raquel Hurtado-Ortiz, Douglas W. Miano, David Nicholson, Guilherme Oliveira, Carlos Ospina Bravo, Fabian Rohden, Ole Seberg, Gernot Segelbacher, Yogesh Shouche, Alejandra Sierra, Ilene Karsch-Mizrachi, Jessica da Silva, Desiree M. Hautea, Manuela da Silva, Mutsuaki Suzuki, Kassahun Tesfaye, Christian Keambou Tiambo, Krystal A. Tolley, Rajeev Varshney, María Mercedes Zambrano & Jörg Overmann
Nature Communications (2022) 13, 1086 DOI:10.1038/s41467-022-28594-0
A diverse group of researchers from around the world have united to demand a sensible international policy solution for “digital sequence information”. In a paper published on in the journal Nature Communications, 41 researchers argue for why a policy solution on digital sequence information (DSI) is urgently needed, and propose a policy framework that would support biodiversity conservation while also providing a realistic mechanism to better share the benefits of DSI research.
Urgent international action is needed to stem the ongoing destruction of our planet’s biodiversity, and the United Nations Convention on Biological Diversity (CBD) is one of the most important global instruments by which such action could be coordinated. Parties to the Convention are currently negotiating the Post-2020 Global Biodiversity Framework which will set the ambition and shape the actions of countries and stakeholders to protect our planet for the coming decades. Disagreements, however, have arisen among Parties to the CBD regarding how to treat (regulate?) data derived from sovereign genetic resources, known as “digital sequence information” or “DSI”. A solution to this issue must be found before an agreement can be reached on the Global Biodiversity Framework.
Scientists have a long history of sharing DSI openly in public databases on the web. This culture of sharing is central to modern biology and biodiversity research, and has helped drive technological advances in fields as diverse as medicine, food security, and green energy production. Public online databases currently contain sequence information for many hundreds of thousands of organisms and grow each day. They support scientific reproducibility, transparency, and advancement. Nonetheless, it is clear that the benefits that arise from this research are unequally distributed around the globe.
In the paper published at Nature Communications, the authors argue that the benefits of DSI can and must be distributed equitably in a way that supports the aims of the CBD without undermining the research community’s cherished culture of sharing. They propose a policy mechanism whereby researchers would continue to deposit and share their data through public online databases, but users of these databases, or high-income nations, would be obliged to contribute to an international fund, which would then distribute monies to countries around the world based on their development status and their contribution to the global DSI dataset, thereby incentivizing countries to generate DSI data on their biodiversity. The result would be a positive feedback loop that shares benefits and incentivizes biodiversity research, without disrupting scientific data sharing. They argue that such a policy mechanism must be “multilateral” to succeed: Nations around the world must cooperate and agree on common rules and that it should be “de-coupled” from access, meaning payments are not connected to viewing the data. The authors of this work also call on policy-makers to engage with researchers in their countries who depend on DSI, so that any policy solution will not hinder crucial biodiversity research.
The authors of this paper are members of the DSI Scientific Network, a group of scientists from different countries and economic settings that share convergent points of view in the DSI debate, and who are joining their voices to argue for sensible policies solutions on this crucial issue.
One of the authors, Mutsuaki Suzuki, Director of NIG INNOVATION at the National of NIG, also founder of ABS Support Team in the NBRP Information Development Program. He has also participated in the Conference of the Parties to the Convention on Biological Diversity as a Japanese government delegate for more than 10 years.

Evolutionary alterations in gene expression and enzymatic activities of gibberellin 3-oxidase 1 in Oryza
Kyosuke Kawai, Sayaka Takehara, Toru Kashio, Minami Morii, Akihiko Sugihara, Hisako Yoshimura, Aya Ito, Masako Hattori, Yosuke Toda, Mikiko Kojima, Yumiko Takebayashi, Hiroyasu Furuumi, Ken-ichi Nonomura, Bunzo Mikami, Takashi Akagi, Hitoshi Sakakibara, Hidemi Kitano, Makoto Matsuoka & Miyako Ueguchi-Tanaka
Communications Biology (2022) 5, 67 DOI:10.1038/s42003-022-03008-5
The plant hormone gibberellin (GA) plays important roles in various developmental events, such as stem elongation, induction of seed germination and flowering. Although GA is indispensable for anther and pollen development, our knowledge of GA functions during plant reproduction has been limited to date.
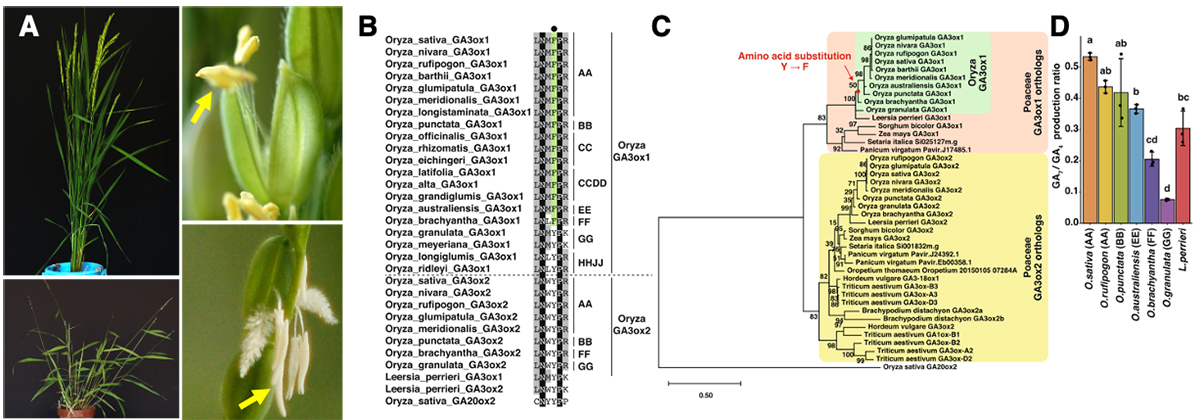
This paper reports the functional and evolutionary analyses of rice gibberellin 3-oxidase 1 (OsGA3ox1), a gibberellin synthetic enzyme specifically expressed in the late developmental stages of anthers. Enzymatic and X-ray crystallography analyses reveal that OsGA3ox1 has a higher GA7 synthesis ratio than OsGA3ox2. In addition, we generate an osga3ox1 knockout mutant by genome editing and demonstrate the bioactive gibberellic acid synthesis by the OsGA3ox1 action during starch accumulation in pollen via invertase regulation. Furthermore, we analyze the evolution of Oryza GA3ox1s and reveal that their enzyme activity and gene expression have evolved in a way that is characteristic of the Oryza genus and contribute to their male reproduction ability.
In this paper, we used the wild strains of genus Oryza that NIG has conserved with the support of National Bioresource Project (NBRP) Rice, Ministry of Education, Culture, Sports, Science and Texhnology (MEXT), Japan.

Professor Akatsuki Kimura has published an English textbook “Quantitative Biology – A practical introduction” from Springer.
The textbook is based on his lecture on quantitative biology at SOKENDAI, including the codes of computer programming.
Target reader is a complete beginner of computer programming.
The textbook also overviews “Cell Architectonics”, a cell biology research Prof. Kimura is pursuing.
Mr. Harsha Somashekar (Plant Cytogenetics/Nonomura Lab), a PhD student (D4) in the graduate university SOKENDAI, received the Best Papers Award 2021 in the 93rd Annual Meeting of the Genetics Society of Japan.

Title:
Hyper accumulation of callose at extracellular spaces of anther locules is required for normal progression of male meiosis in rice.
The auxin-inducible degron 2 (AID2) system enables controlled protein knockdown during embryogenesis and development in Caenorhabditis elegans.
Negishi T#, Kitagawa S#, Horii N, Tanaka Y, Haruta N, Sugimoto A, Sawa H, Hayashi KI, Harata M*, Kanemaki MT*.
# These authors contributed equally * Co-corresponding authors
Genetics (2022) 220, iyab218 DOI:10.1093/genetics/iyab218
To analyze the protein function of C. elegans, it is useful to study the phenotype by suppressing the protein function. For this purpose, nematode individuals with mutated genes and the RNA interference method have been used. However, genes that play an essential role in nematode development may cause a developmental defect due to the loss of the gene, making further analysis difficult. In addition, due to a large amount of maternally-derived mRNA and proteins in the eggs, the functions of proteins normally involved in early development may not be expressed as a phenotype in early development using existing techniques. The auxin-inducible degron (AID) system that we have established rapidly degrades and removes target proteins at any given time and allows us to observe phenotypes that cannot be seen using existing techniques. The AID method has already been applied to C. elegans by a group in the U.S. and is now being used in various studies. However, the conventional AID method has some problems, such as leaky target degradation in the absence of auxin and the effects of high concentrations of auxin.
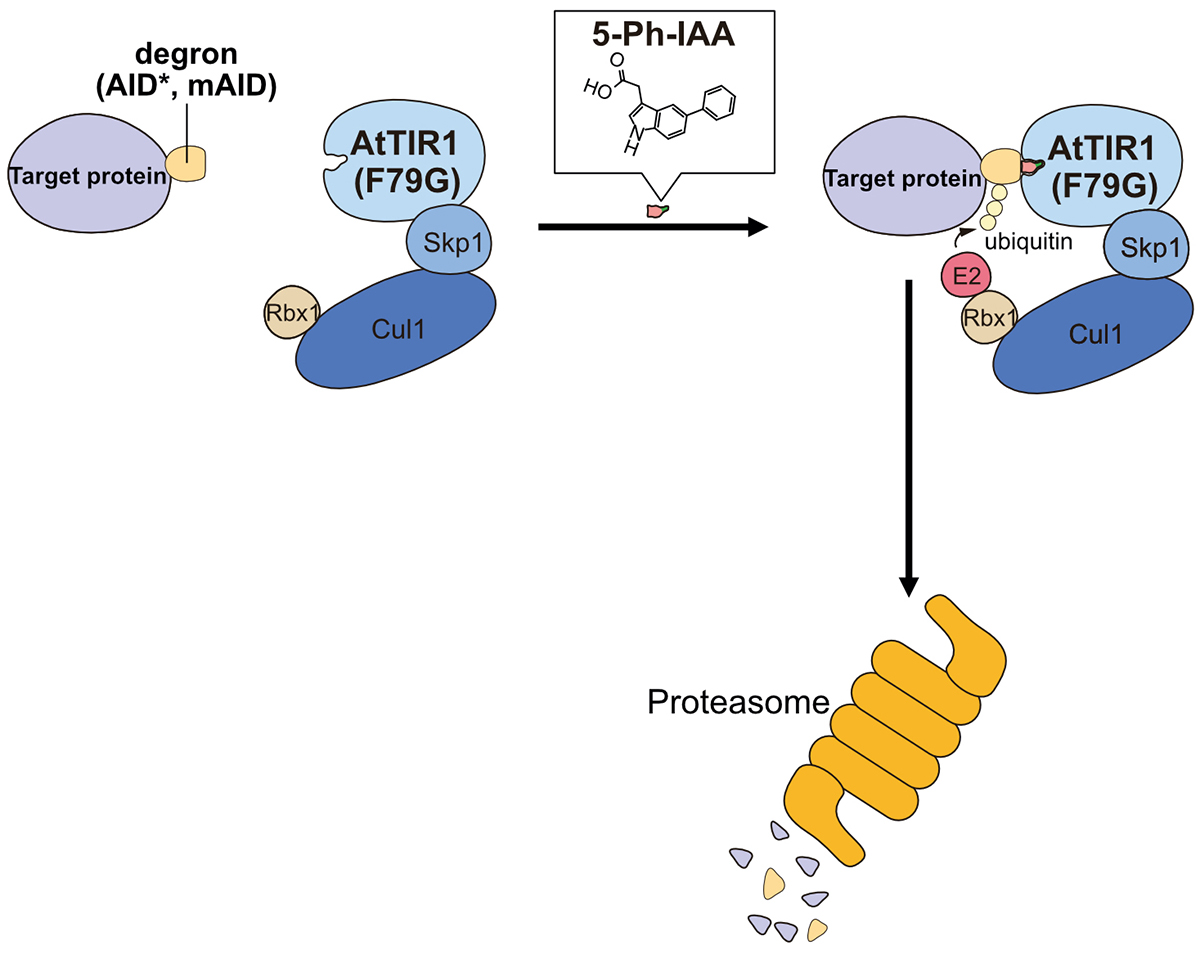
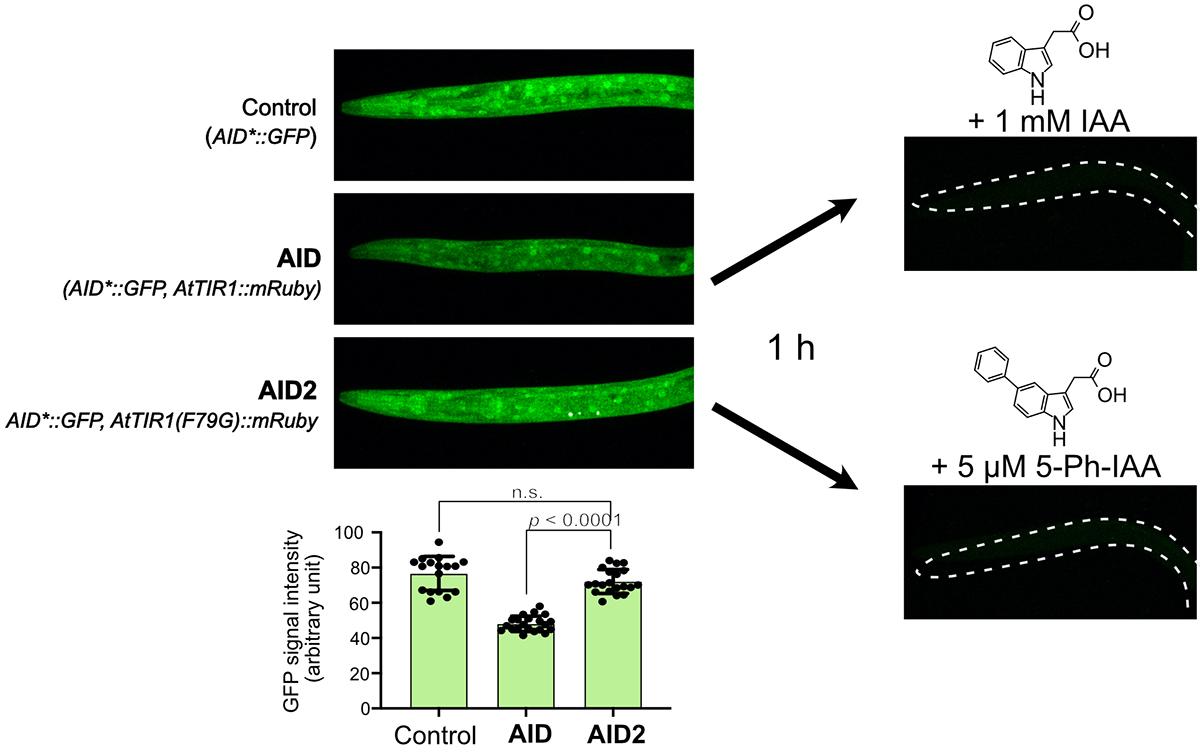
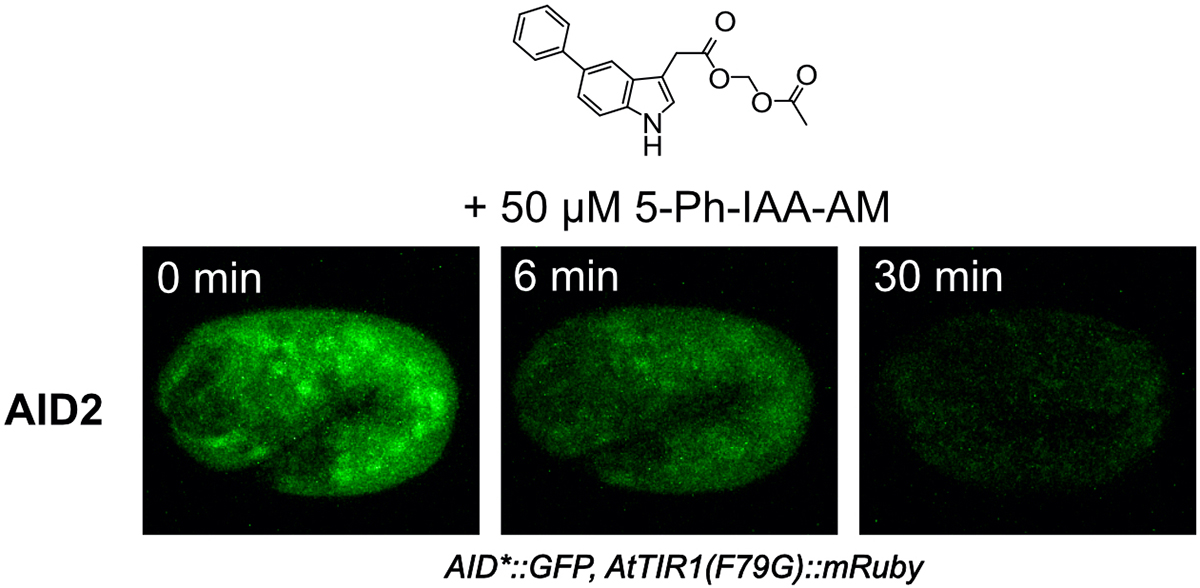
Therefore, we applied an improved method, AID2, which we developed last year using budding yeast, cultured cells, and mice, to C. elegans to overcome these problems (Figure 1). As a result, we found that AID2 completely suppressed the leaky degradation in C. elegans and rapidly induced target degradation with 1/1300 of the ligand concentration (Figure 2). Furthermore, by degrading the histone H2A.Z protein, we succeeded in observing a developmental defect that had not been reported previously. Furthermore, to induce degradation in the embryo in the egg, we developed a modified ligand suitable for eggshell permeabilization, enabling rapid proteolysis in the embryo (Figure 3).
This research was led by Assistant Professor Takefumi Negishi and Professor Masato Kanemaki at the National Institute of Genetics, and a graduate student Saho Kitagawa and Professor Masahiko Harada at Tohoku University, in collaboration with Professor Kenichiro Hayashi at Okayama University of Science, Professor Hitoshi Sawa at the National Institute of Genetics, and Professor Asako Sugimoto at Tohoku University.
Ligand-induced degrons for studying nuclear functions
Masato T. Kanemaki
Current Opinion in Cell Biology, advanced online publication (2022) 74, 29–36 DOI:10.1016/j.ceb.2021.12.006
Nuclear functions such as transcription, DNA replication, and DNA repair are closely related to cell proliferation and chromosome segregation. Since cultured cells usually double in about 24 hours, it is crucial to remove the target protein within a few minutes to a few hours to investigate its function in order to avoid secondary effects (Fig. 1). Therefore, the “degron methods”, which enable rapid depletion of a target protein, are ideal for studying nuclear functions.
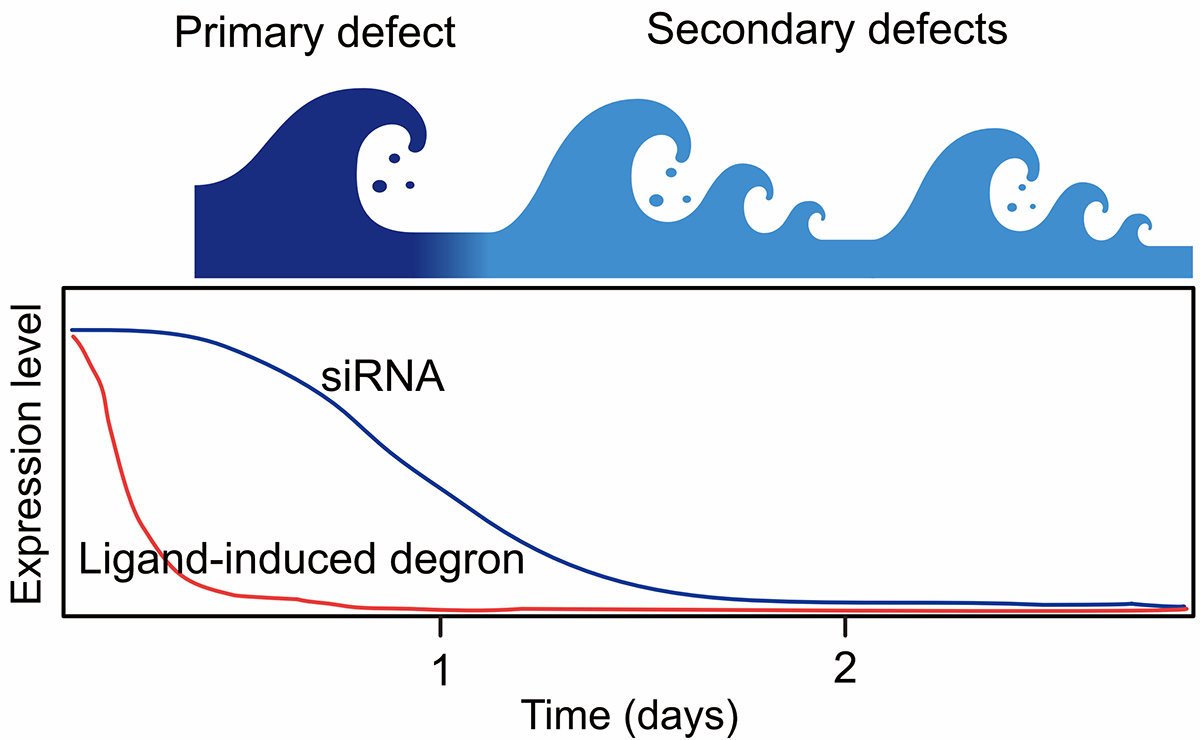
Figure1: Comparison of the effects of target protein when using degron and siRNA methods
In this review article, we described the degron technologies developed so far, including the auxin degron (AID) method developed by our laboratory, and introduced how they have been helpful for research on nuclear functions (Fig. 2). Since degron technologies are relatively new research methods, they are expected to be useful for many studies in the future.
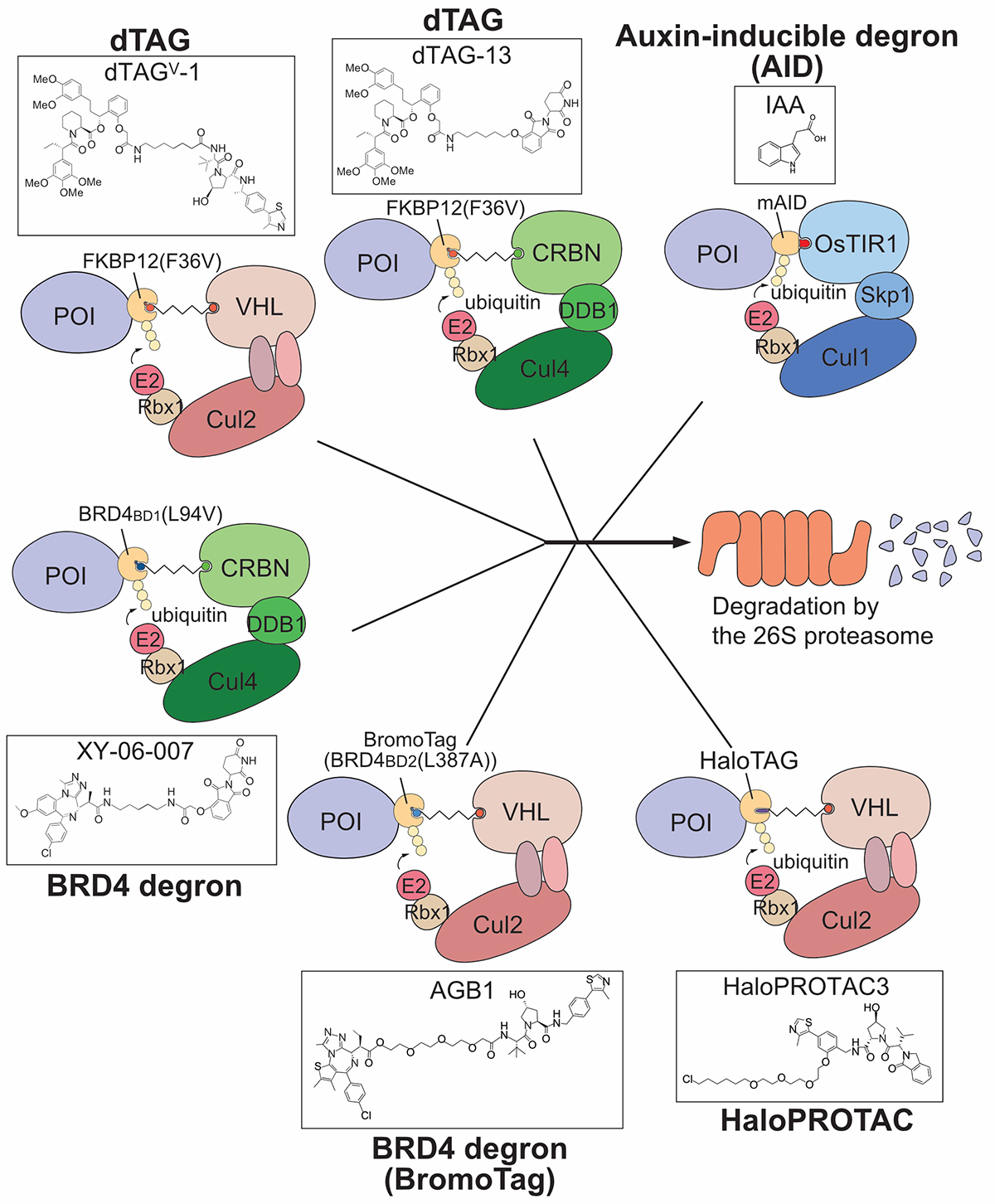
Figure2: The ligand-dependent degron methods developed to date. Except for AID, they are based on a pharmaceutical technology, PROTAC.
This paper will be published in the “Cell Nucleus” issue (June, 2022) in Current Opinion in Cell Biology, edited by Prof. Kazuhiro Maeshima at National Institute of Genetics and Prof. Eran Meshorer at the Hebrew University of Jerusalem.










