Press release
Comprehensive discovery of CRISPR-targeted terminally redundant sequences in the human gut metagenome: viruses, plasmids, and more
R. Sugimoto, L. Nishimura, P. T. Nguyen, J. Ito, N. F. Parrish, H. Mori, K. Kurokawa, H. Nakaoka, I. Inoue
PLOS Computational Biology (2021) 17, e1009428 DOI:10.1371/journal.pcbi.1009428
![]() Press release (In Japanese only)
Press release (In Japanese only)
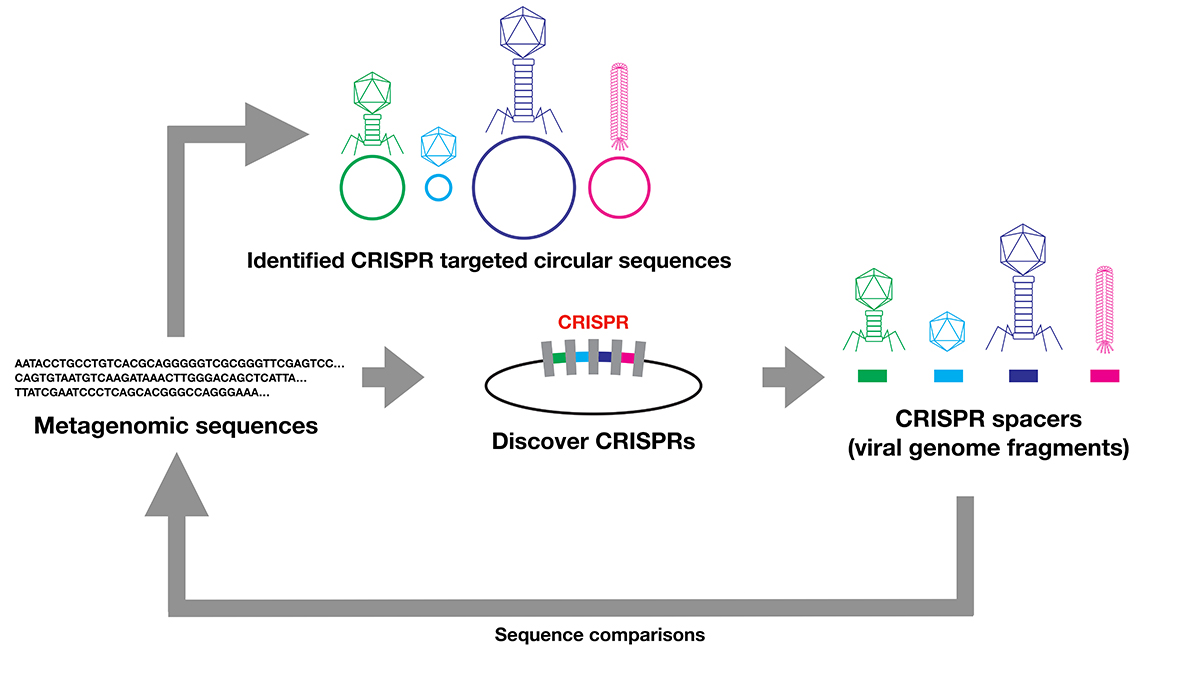
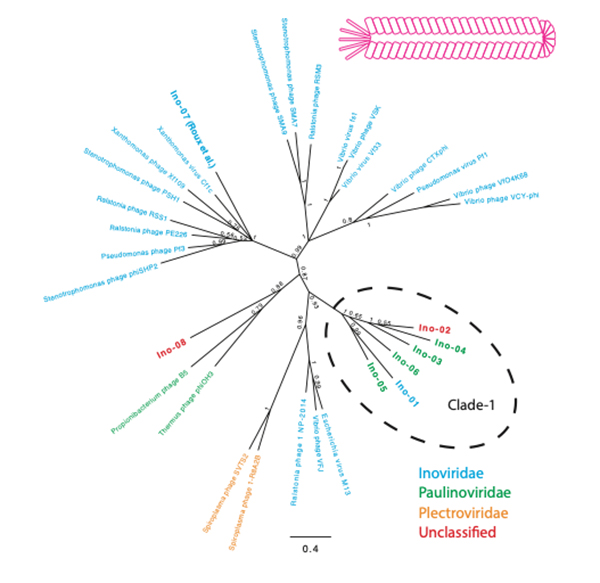
The evolution and origins of viruses are long-standing questions in the field of biology. Viral genomes provide fundamental information to infer the evolution and origin of viruses. However, viruses are extraordinarily diverse, and there are no single genes shared across entire species. Several methods were developed to collect viral genomes from metagenome. To infer viral genomes from metagenome, previous approaches relied on reference viral genomes. We thought that such reference-based methods may not be sufficient to uncover diverse viral genomes; therefore, we developed a pipeline that utilizes CRISPR, a prokaryotic adaptive immunological memory. Using this pipeline, we discovered more than 10,000 positively complete CRISPR-targeted genomes from human gut metagenome datasets. A substantial portion of the discovered genomes encoded various types of capsid proteins, supporting the contention that these sequences are viral. Although the majority of these capsid-protein-coding sequences were previously characterized, we notably discovered Inoviridae genomes that were previously difficult to infer as being viral. Furthermore, some of the remaining unclassified sequences without a detectable capsid-protein-encoding gene had a notably low protein-coding ratio. Overall, our pipeline successfully discovered viruses and previously uncharacterized presumably mobile genetic elements targeted by CRISPR.
Source: R. Sugimoto et al., PLOS Computational Biology DOI:10.1371/journal.pcbi.1009428