



Saga Group / Mammalian Development Laboratory
Formal proof of the requirement of MESP1 and MESP2 in mesoderm specification and their transcriptional control via specific enhancers in mice
Rieko Ajima, Yuko Sakakibara, Noriko Sakurai-Yamatani, Masafumi Muraoka and Yumiko Saga
Development (2021) 148, dev194613 DOI:10.1242/dev.194613
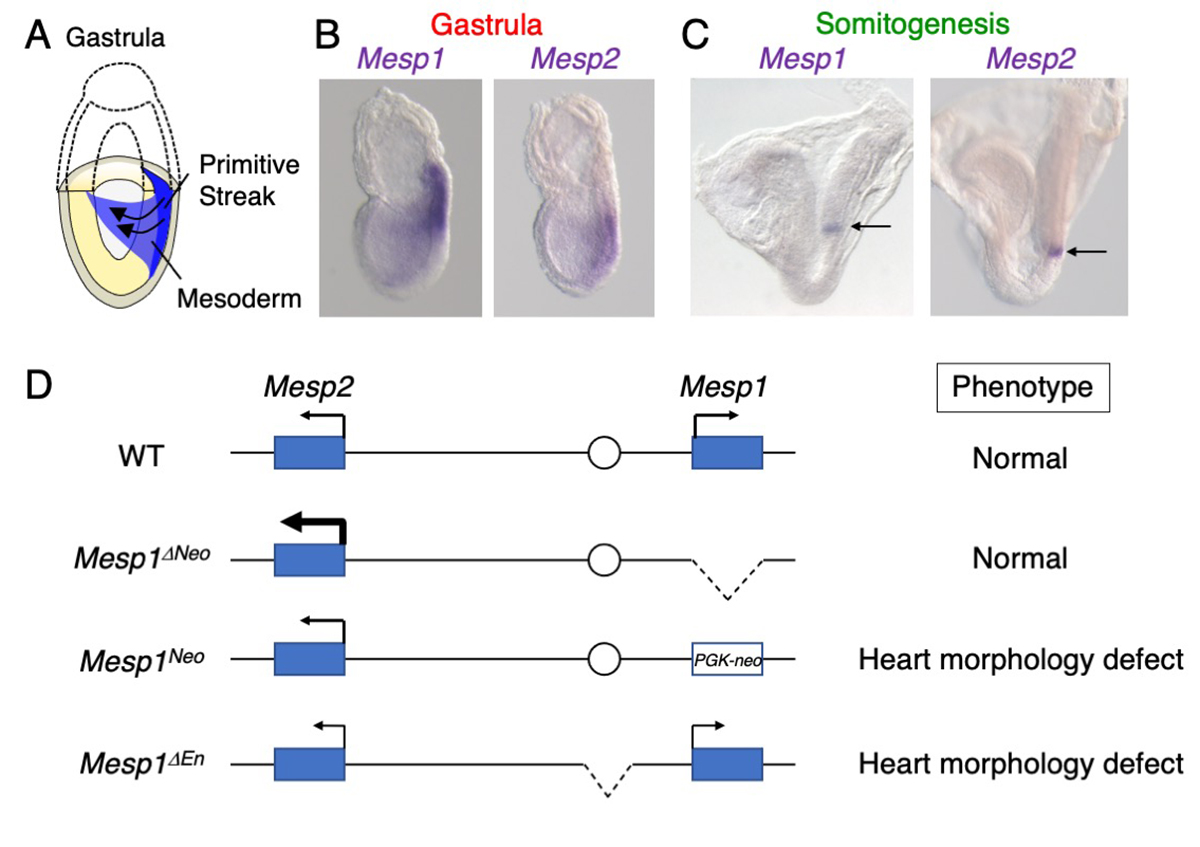
The mesodermal cells are derived from the primitive streak during gastrulation in mouse development (Figure A). These mesodermal cells give rise to the heart, somites, kidney, and limbs. MESP1 and MESP2 are transcriptional factors expressed in early mesoderm and boundary of newly forming somites (Figure B, C). The Mesp1/Mesp2 dKO embryos exhibited markedly severe mesoderm formation defects. However, MESP1 and MESP2 were thought to regulate distinct targets, because Mesp1 and Mesp2 single KO embryos display distinct phenotypes, heart morphogenesis defects and somite formation defects, respectively.
In this study, we established the Mesp1/Mesp2 mutants using genome editing techniques, and found the Mesp1/Mesp2 dKO embryos exhibited defects similar to the original Mesp1/Mesp2 dKO embryos. However, Mesp1 KO did not display any phenotypes. We noted up-regulation of Mesp2 in the Mesp1 KO embryos, suggesting that MESP2 rescues the loss of MESP1 in mesoderm specification. We also found that Mesp1 and Mesp2 expression in the early mesoderm is regulated by the common enhancer. Deletion of the enhancer caused the down-regulation of both genes, resulting in heart formation defects. This study suggests dosage-dependent roles of MESP1 and MESP2 in early mesoderm formation.
Press release
Comprehensive discovery of CRISPR-targeted terminally redundant sequences in the human gut metagenome: viruses, plasmids, and more
R. Sugimoto, L. Nishimura, P. T. Nguyen, J. Ito, N. F. Parrish, H. Mori, K. Kurokawa, H. Nakaoka, I. Inoue
PLOS Computational Biology (2021) 17, e1009428 DOI:10.1371/journal.pcbi.1009428
![]() Press release (In Japanese only)
Press release (In Japanese only)
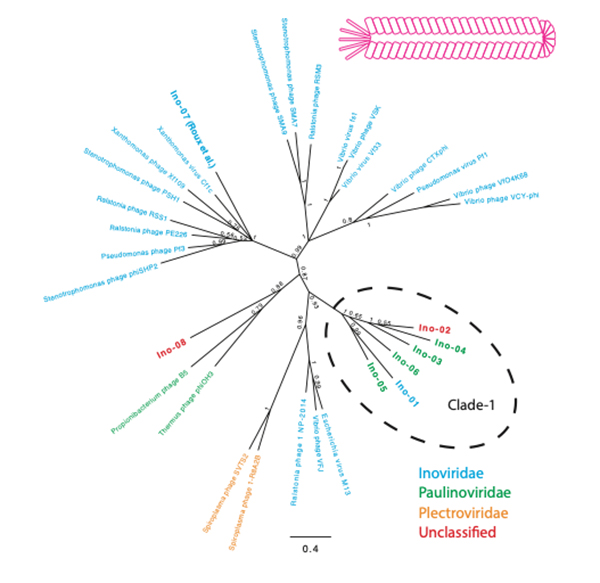
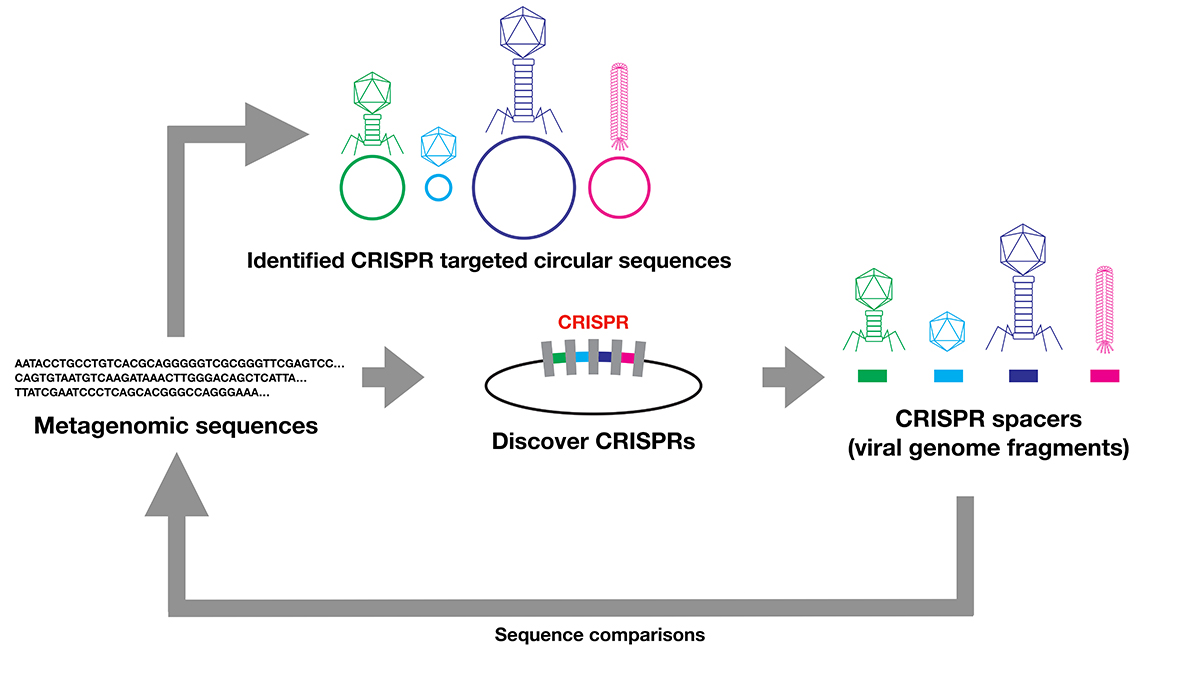
The evolution and origins of viruses are long-standing questions in the field of biology. Viral genomes provide fundamental information to infer the evolution and origin of viruses. However, viruses are extraordinarily diverse, and there are no single genes shared across entire species. Several methods were developed to collect viral genomes from metagenome. To infer viral genomes from metagenome, previous approaches relied on reference viral genomes. We thought that such reference-based methods may not be sufficient to uncover diverse viral genomes; therefore, we developed a pipeline that utilizes CRISPR, a prokaryotic adaptive immunological memory. Using this pipeline, we discovered more than 10,000 positively complete CRISPR-targeted genomes from human gut metagenome datasets. A substantial portion of the discovered genomes encoded various types of capsid proteins, supporting the contention that these sequences are viral. Although the majority of these capsid-protein-coding sequences were previously characterized, we notably discovered Inoviridae genomes that were previously difficult to infer as being viral. Furthermore, some of the remaining unclassified sequences without a detectable capsid-protein-encoding gene had a notably low protein-coding ratio. Overall, our pipeline successfully discovered viruses and previously uncharacterized presumably mobile genetic elements targeted by CRISPR.
Source: R. Sugimoto et al., PLOS Computational Biology DOI:10.1371/journal.pcbi.1009428
Maeshima Group / Genome Dynamics Laboratory
Telomere-specific chromatin capture using a pyrrole–imidazole polyamide probe for the identification of proteins and non-coding RNAs
Satoru Ide#*, Asuka Sasaki#, Yusuke Kawamoto, Toshikazu Bando, Hiroshi Sugiyama, Kazuhiro Maeshima
#Equally contributed, *Corresponding author
Epigenetics & Chromatin (2021) 14, 46 DOI:10.1186/s13072-021-00421-8
Background: Knowing chromatin components at a DNA regulatory element at any given time is essential for understanding how the element works during cellular proliferation, differentiation and development. A region-specific chromatin purification is an invaluable approach to dissecting the comprehensive chromatin composition at a particular region. Several methods (e.g., PICh, enChIP, CAPTURE and CLASP) have been developed for isolating and analyzing chromatin components. However, all of them have some shortcomings in identifying non-coding RNA associated with DNA regulatory elements.
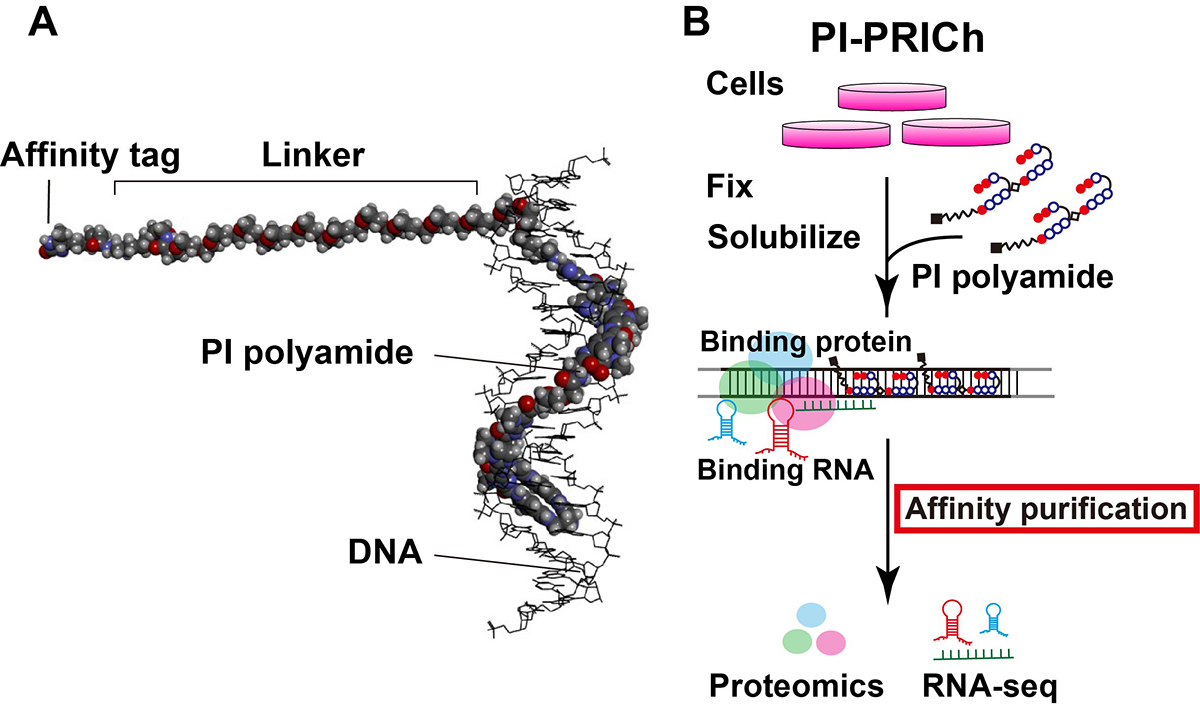
Results: We have developed a new approach for affinity purification of specific chromatin segments employing an N-methyl pyrrole (P)−N-methylimidazole (I) (PI) polyamide probe, which binds to a specific sequence in double-stranded DNA via Watson–Crick base pairing as a minor groove binder (Figure 1A). This new technique is called proteomics and RNA-omics of isolated chromatin segments (PI-PRICh). Using PI-PRICh to isolate mouse and human telomeric components, we found enrichments of shelterin proteins, the well-known telomerase RNA component (TERC) and telomeric repeat-containing RNA (TERRA) When PI-PRICh was performed for alternative lengthening of telomere (ALT) cells with highly recombinogenic telomeres, in addition to the conventional telomeric chromatin, we obtained chromatin regions containing telomeric repeat insertions scattered in the genome and their associated RNAs.
Conclusion: PI-PRICh reproducibly identified both the protein and RNA components of telomeric chromatin when targeting telomere repeats. PI polyamide is a promising alternative to simultaneously isolate associated proteins and RNAs of sequence-specific chromatin regions under native conditions, allowing better understanding of chromatin organization and functions within the cell (Figure 1B).
This work was supported by an NIG-JOINT (2015-B6), JSPS grants (JP17J10836 to A.S.; 15H01361 and 21H02535 to S.I.; 20H05936 and 21H02453 to K.M.), the Takeda Science Foundation to K.M. and the Uehara Memorial Foundation to K.M.. A.S. was a JSPS Fellow (DC2).
Arita Group / Biological Networks Laboratory
A sugar utilization phenotype contributes to the formation of genetic exchange communities in lactic acid bacteria
Shinkuro Takenaka, Takeshi Kawashima, Masanori Arita.
FEMS Microbiology Letters (2021) 368, fnab117 DOI:10.1093/femsle/fnab117
In prokaryotes, a major contributor to genomic evolution is the exchange of genes via horizontal gene transfer (HGT). Areas with a high density of HGT networks are defined as genetic exchange communities (GECs). Although some phenotypes associated with specific ecological niches are linked to GECs, little is known about the phenotypic influences on HGT in bacterial groups within a taxonomic family. Thanks to the published genome sequences and phenotype data of lactic acid bacteria (LAB), it is now possible to obtain more detailed information about the phenotypes that affect GECs. Here, we have investigated the relationship between HGT and internal and external environmental factors for 178 strains from 24 genera in the Lactobacillaceae family. We found a significant correlation between strains with high utilization of sugars and HGT bias. The result suggests that the phenotype of the utilization of a variety of sugars is key to the construction of GECs in this family. This feature is consistent with the fact that the Lactobacillaceae family contributes to the production of a wide variety of fermented foods by sharing niches such as those in vegetables, dairy products and brewing-related environments. This result provides the first evidence that phenotypes associated with ecological niches contribute to form GECs in the LAB family.
Source: S. Takenaka, et al., DOI: 10.1093/femsle/fnab117
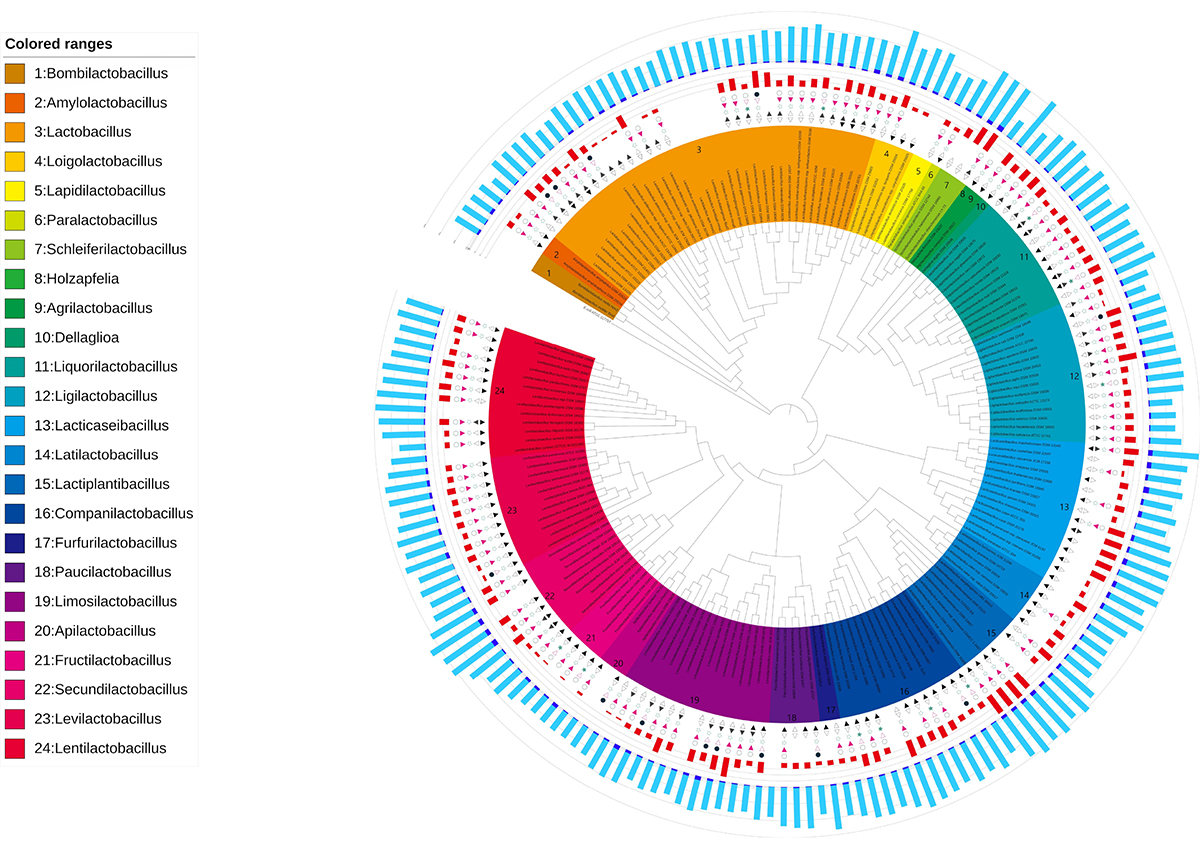
Figure at the beginning: Phylogenetic tree based on the 16S rRNA genes of the LAB strains with the phenotypic and genomic features identified. The inner band shows species colored by genus. The next five symbols show phenotypic characteristics for each LAB strain; first inward-facing triangle indicates the growth at 15°C, second outward-facing triangle indicates the growth at 45°C, third star indicates the micro aerophilic, fourth red inward-facing indicates facultatively anaerobic and fifth circle indicates obligate anaerobic. A filled symbol means the strain has the phenotype, and an open symbol means that it does not. A blank means that there is no relevant information available. The next red band shows the number of sugar types that can be utilized. The outer bands show the number of coding sequences (CDS) for each strain: navy blue indicates the estimated number of CDS acquired by the horizontal gene transfer (HGT) and light blue indicates the number of native CDS.
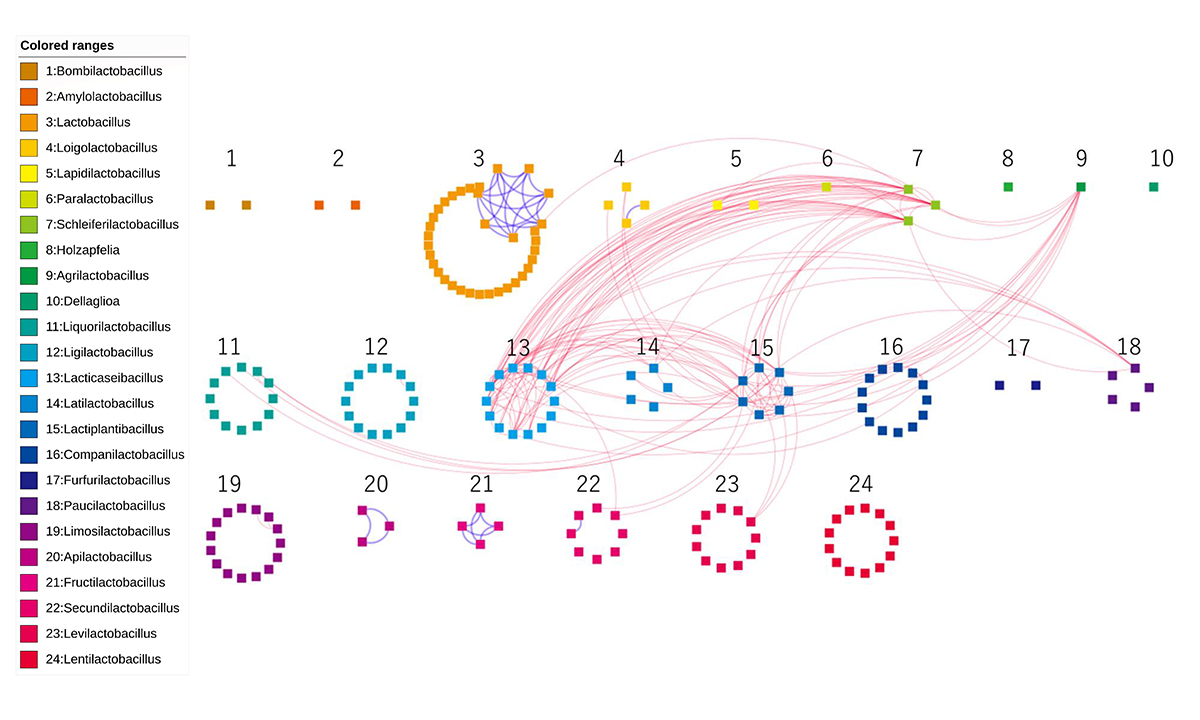
Figure: The networks for the generalist and specialist group orthologs. Each of the 178 nodes represents an LAB genome, which are colored and numbered by genus. Edges of dotted-red/solid-blue were created between two genomes when the number of sharing generalist/specialist group orthologs was more than five.
Dr. Keisuke Yonehara joined NIG as a professor on October 1.
YONEHARA, Keisuke : Multiscale Sensory Structure Laboratory











