



3D genomics across the tree of life reveals condensin II as a determinant of architecture type
Hoencamp, C., Dudchenko, O., Elbatsh, A. M.O., Brahmachari, S., Raaijmakers, J. A., van Schaik, T., Cacciatore, Á. S., Contessoto, V., van Heesbeen, R. G.H.P., van den Broek, B., Mhaskar, A. N., Teunissen, H., St Hilaire, B. G., Weisz, D., Omer, A. D., Pham, M., Colaric, Z., Yang, Z., Rao, S. S.P., Mitra, N., Lui, C., Yao, W., Khan, R., Moroz, L. L., Kohn, A., St. Leger, J., Mena, A., Holcroft, K., Gambetta, M. C., Lim, F., Farley, E., Stein, N., Haddad, A., Chauss, D., Mutlu, A. S., Wang, M. C., Young, N. D., Hildebrandt, E., Cheng, H. H., Knight, C. J., Burnham, T. L.U., Hovel, K. A., Beel, A. J., Mattei, P.-J., Kornberg, R. D., Warren, W. C., Cary, G., Gómez-Skarmeta, J. L., Hinman, V., Lindblad-Toh, K., di Palma, F., Maeshima, K., Multani, A. S., Pathak, S., Nel-Themaat, L., Behringer, R. R., Kaur, P., Medema, R. H., van Steensel, B., de Wit, E., Onuchic, J. N., Di Pierro, M., Lieberman-Aiden, E., Rowland, B. D.
Science 372, 984-989 (2021) DOI:10.1126/science.abe2218
An international collaborative team, which was led by B.D. Rowland at the Netherlands Cancer Institute and E. Lieberman Aiden (NIG International Strategic Advisor) at Baylor College of Medicine, investigated genome folding across the eukaryotic tree of life (24 species, Fig. B). The team found four types of 3D genome architecture at chromosome-scale (Fig. A). Each type appeared and disappeared repeatedly during eukaryotic evolution. The type of genome architecture that an organism exhibits correlates with the absence of condensin II subunits (Fig. B). Condensin is a protein complex required for mitotic chromosome assembly. In vertebrates, it is known that there are two types of condensins, condensin I and II, and that condensin II shortens mitotic chromosomes.
Moreover, the team demonstrated that condensin II depletion converted the architecture of the human genome to a state resembling that seen in organisms such as fungi or mosquitoes (Rabl-like, Fig. C). In this state, centromeres clustered together at nucleoli, and heterochromatin domains merged. The team proposed a physical model in which lengthwise compaction of chromosomes by condensin II during mitosis determines chromosome-scale genome architecture, with effects that are retained during the subsequent interphase. This mechanism is likely conserved since the last common ancestor of all eukaryotes.
The part to which NIG contributed was supported by MEXT KAKENHI grants (20H05936).
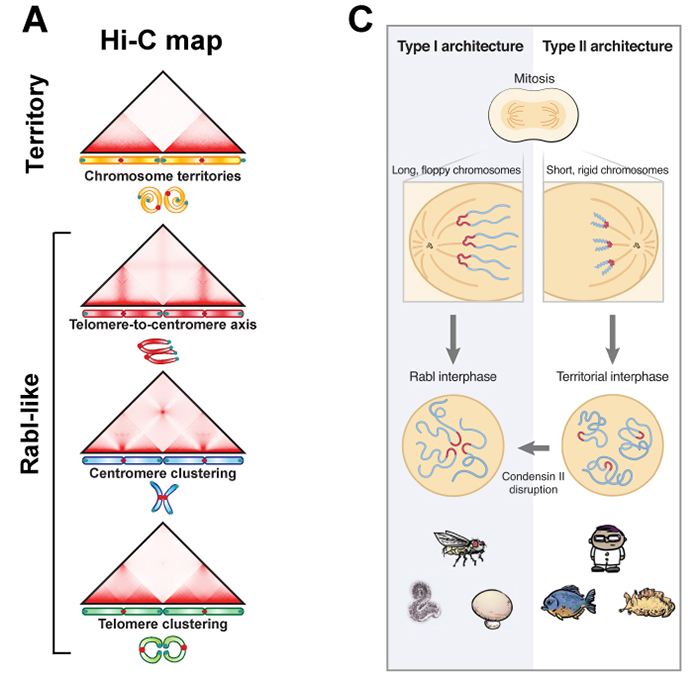
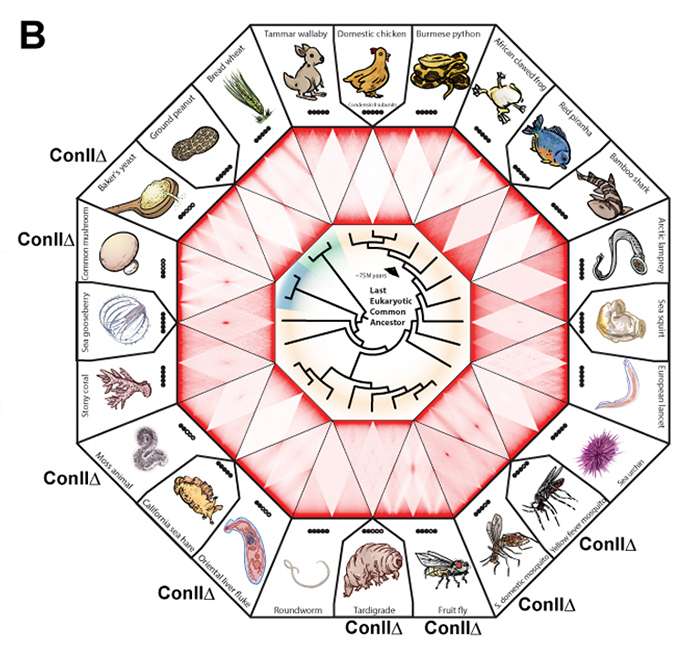
Figure: (A) Chromosome positioning in the nucleus classified by Hi-C maps: “chromosome territory” and “rabl-like”. (B) Hi-C maps of 24 species covering animals (yellow), fungi (blue) and plants (green). Presence of the condensin II subunits in each species is indicated by solid black circles. In case of condensin II-deficiency, shown as “Con IIΔ”. (C) Model. Having shorter chromosomes during cell division leads to separate centromeres and territorial genome architecture in the subsequent interphase. Reducing chromosome lengthwise compaction by condensin II disruption, leads to enhanced centromere clustering, loss of chromosome territories, and a Rabl-like genome architecture.
Our website will be under maintenance from 3:00 p.m. on May 27 to 9:00 a.m. on May 28. The website will be basically available during the maintenance period, but may be intermittently unavailable.
Thank you for your understanding and cooperation.
本日5月27日15:00より翌28日9:00まで、遺伝研ウェブサイトのメンテナンスを予定しています。メンテナンス時間においてもウェブサイトは利用できますが、断続的に利用できない場合があります。
皆様のご理解とご協力をお願いいたします。
Press release
NeuroGT: A brain atlas of neurogenic tagging CreER drivers for birthdate-based classification and manipulation of mouse neurons
T Hirata*, Y Tohsato, H Itoga, G Shioi, H Kiyonari, S Oka, T Fujimori, S Onami *Corresponding author
Cell Reports Methods 1, 100012 (2021) DOI:10.1016/j.crmeth.2021.100012
Press release (In Japanese only)
Neuronal birthdate is one of the major determinants of neuronal phenotypes. However, most birthdating methods are retrospective in nature, allowing very little experimental access to the classified neuronal subsets. Hirata and her collaborators have developed four neurogenic tagging mouse lines that can assign CreER-loxP recombination to neuron subsets that share the same differentiation timing in living animals. Because this genetic tag is irreversible, the classified neuronal subset can be subsequently subjected to various experimental manipulations.
To encourage the use of this resource, Hirata et al. have launched “NeuroGT database”, a brain atlas of neurogenic tagging mouse lines, which includes holistic image data of the loxP-recombined neurons and their processes across the entire brain that were tagged at each single day during the neurodevelopmental period. Users can search for the datasets from a web browser using the terns in the meta-information, interactively view thumbnail images of the sections, and download the high-resolution section images.
The NeuroGT is now open to public, offering people the opportunity to find specific neurogenic tagging driver lines and the stages of tagging appropriate for their own research purposes. The driver mouse lines can be obtained from Riken Bioresource Center.
The NeuroGT database was constructed by joint efforts of different research groups; mouse engineering (RIKEN Center for Biosystems Dynamics Research), neuroscience (National Institute of Genetics), bioimaging (National Institute for Basic Biology), and image informatics (Ritsumeikan University, RIKEN Center for Biosystems Dynamics Research)
This research was supported by ROIS Challenging Exploratory Research Projects for the Future Grant and Grant-in-Aid for Publication of Scientific Research Results (Database, 19HP7002). High resolution digitization of section images was supported by Advanced Bioimaging Support (JP16H06280) and Grants-in-Aid for Scientific Research (20H03345, JP18H05412).
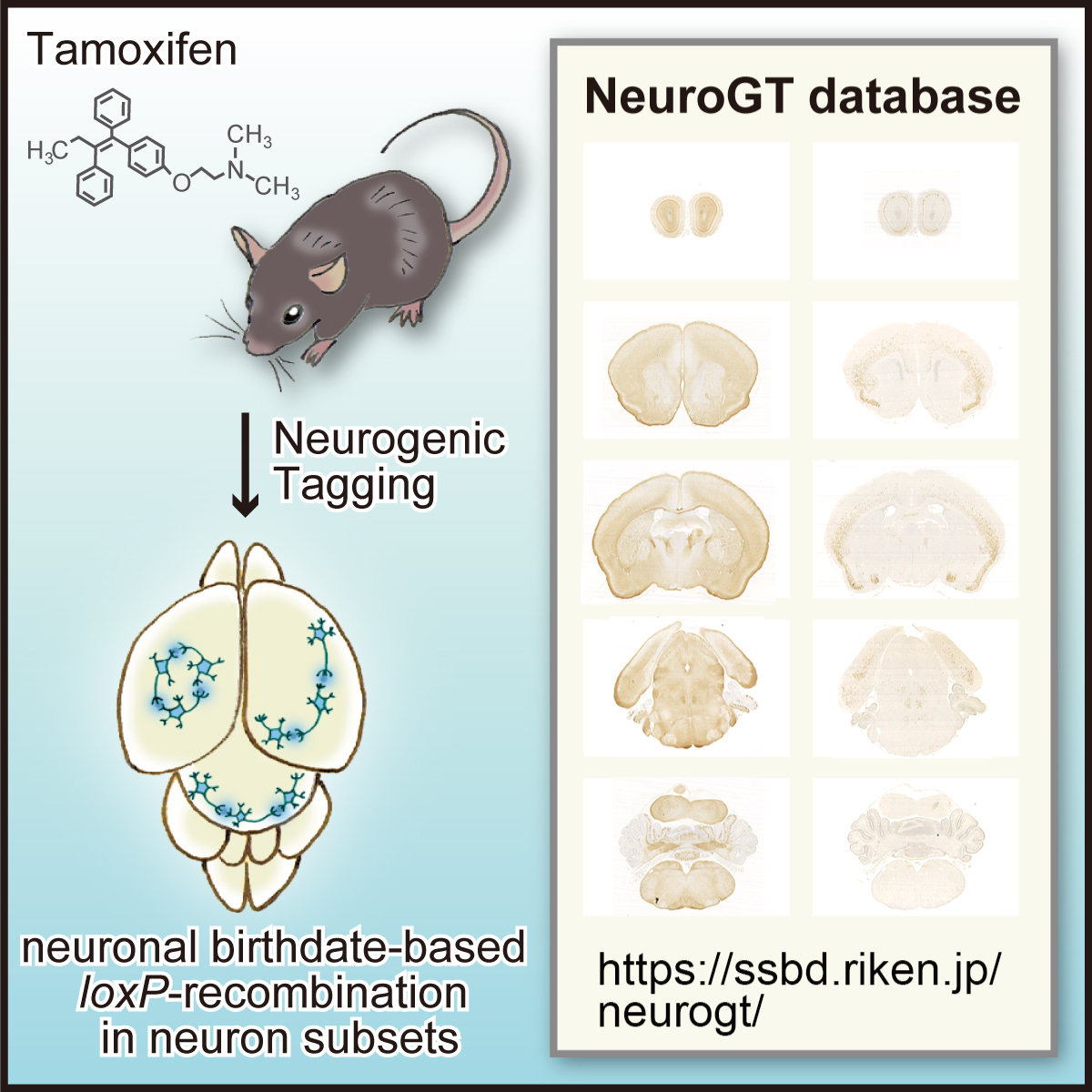
Figure: Graphical abstract of “NeuroGT Database”
Video: Thumbnail images of coronal sections can be sequentially viewed along the antero-posterior brain axis by dragging the slider. The images stained for the two different reporters, membrane-localized GFP and nucleus-localized-βGAL are stacked separately. The sync button automatically matches the section level of the two reporter images.
National Institute of Genetics has offered cooperation to Shizuoka prefecture and has signed a contract concerning investigation cooperation since April 2020. In response to the spread of infection of the new coronavirus variant in these days, a memorandum of cooperation on molecular epidemiological investigation (SARS-CoV-2 RNA whole genome analysis) of novel coronavirus has been signed between our institute and Heita Kawakatsu, Governor of Shizuoka Prefecture, at the government office.
The principles set in the memorandum are: “to support for proactive epidemiological investigation (SARS-CoV-2 RNA whole genome analysis) promoted by Shizuoka prefecture”, “to contribute to treatment of patients infected with novel coronavirus and prevention of the spread of the virus”, “to disclose molecular epidemiological information obtained from specimen samples in order to contribute to the conquest of COVID-19” and “to give due consideration to the protection of personal information when molecular epidemiological information are disclosed”. Based on this memorandum, our institute implements support for SARS-CoV-2 RNA whole genome analysis in earnest.
April 30th, 2021
National Institute of Genetics

Left, Heita Kawakatsu (Governor, Shizuoka Prefecture): Right, Fumio Hanaoka (Director-General, National Institute of Genetics)
Decoding the transcriptome of pre-granulosa cells during the formation of primordial follicles in the mouse
Kurumi Fukuda, Masafumi Muraoka, Yuzuru Kato, and Yumiko Saga
Biology of Reproduction 2021 April 13 DOI:10.1093/biolre/ioab065
6. Primordial follicles, a finite reservoir of eggs in mammalian ovaries, are composed of a single oocyte and its supporting somatic cells, termed granulosa cells. Although their formation may require reciprocal interplay between oocytes and pre-granulosa cells, precursors of granulosa cells, little is known about the underlying mechanisms. We addressed this issue by decoding the transcriptome of pre-granulosa cells during the formation of primordial follicles. We found that marked gene expression changes, including extracellular matrix, cell adhesion, and several signaling pathways, occur along with primordial follicle formation. Importantly, differentiation of pre-granulosa cells to granulosa cells was delayed in mutant ovaries of the germ cell-specific genes Nanos3 and Figla, accompanied by perturbed gene expression in mutant pre-granulosa cells. These results suggest that proper development of oocytes is required for the differentiation of pre-granulosa cells.
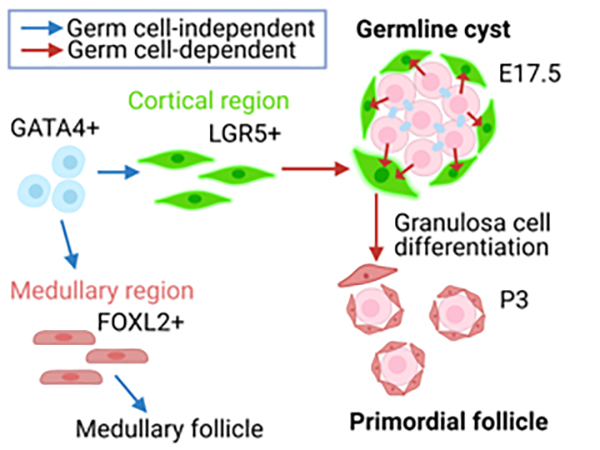
Figure: Pre-granulosa cells (Lgr5+) originate from GATA4 + precursors (blue arrow) in an oocyte-independent manner. On the other hand, differentiation of pre-granulosa cells to granulosa cells requires oocytes (red arrow).










