



Division of Molecular and Developmental Biology・Kawakami Group
The spatio-temporal regulation of muscle contractions that generate body movements critically depends on the exquisitely precise innervation of each muscle type by the appropriate motoneuron subtype. Therefore, delineating the identities of motoneurons and their connectivity to target muscles is fundamental to an understanding of the motor control by the central nervous system. In this study, by taking advantage of the optical and genetic accessibility, we dissected the spinal cord motor column of zebrafish larvae at the cellular level. By using the BAC for the Mnx homeodomain gene mnr2b, we established the mnGFF7 transgenic line expressing the Gal4FF transcription factor in a large part of the motor column. Single cell labelling of Gal4FF-expressing cells in the mnGFF7 larvae enabled a detailed investigation of the morphological characteristics of individual spinal motoneurons, as well as the overall organisation of the motor column in a spinal segment. The transgenic fish established here should facilitate an understanding of the cellular and molecular architecture of the spinal cord motor column and its connection to muscles in vertebrates.
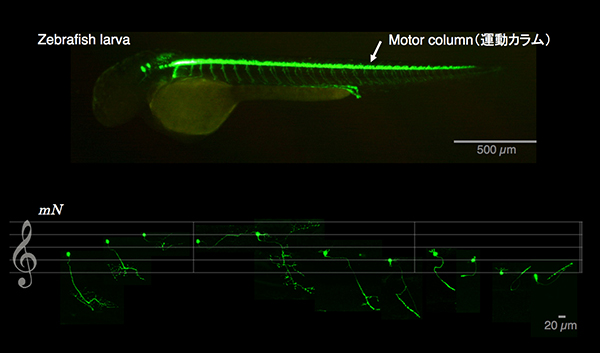
Lateral view of a zebrafish larva (top). Eleven different types of spinal motoneurons identified in the single cell labeling experiment.
Press release
The ancestor of extant Japanese fancy mice contributed to the mosaic genomes of classical inbred strains
Takada, T., Ebata, T., Noguchi, H., Keane, K., Adams, D., Narita, T., Shin-I, T., Fujisawa, H., Toyoda, A., Abe, K., Obata, Y., Sakaki, Y., Moriwaki, K., Fujiyama, A., Kohara, Y. and Shiroishi, T.We resequenced the genomes of Mus musculus molossinus-derived two inbred strains, MSM/Ms and JF1/Ms. MSM/Ms originated from Japanese wild mice, and ancestry of JF1/Ms was originally found in Europe and then transferred to Japan. We compared the characteristics of these sequences to those of the C57BL/6J reference sequence and the recent datasets from the resequencing of 17 inbred strains in the Mouse Genome Project.
The major outcomes of this study are summarized below.
1. Over 10 million SNPs and 1 million short indels are identified between the MSM/Ms or JF1/Ms sequence and the B6 reference sequence.
2. In comparison with the B6 reference sequence, the MSM and JF1 sequences contain 38,182 and 38,124 non-synonymous SNPs in 11,489 and 11,313 genes, respectively.
3. Genome introgression from M. m. molossinus into M. m. domesticus is primary framework for the mosaic genomes of classical inbred strains.
4. The genomes of B6 and other classical inbred strains have long consecutive segments with extremely high similarity (>99.998%) to the JF1/Ms strain. This indicates that the ancestor of JF1/Ms was direct origin of M. m. molossinus genome in classical inbred strains.
5. Roughly 30 to 40% of the SNPs detected in pairwise comparisons of classical inbred strains are attributable to the M. m. molossinus genome introgression.
The sequence data of MSM/Ms and JF1/Ms are available through the NIG mouse genome database, with side-by-side comparison to the B6 reference sequence.
This work was carried out in collaboration with National Institute of Genetics (Mammalian Genetics Laboratory, Comparative Genomics Laboratory and Genome Biology Laboratory), RIKEN (BioResource Center and Genome Science Center) and the Wellcome Trust Sanger Institute, and was supported by a Grant-in-Aid for Scientific Research on Priority Areas “Comparative Genomics” and the National BioResource Projects “the Genome Information Upgrading Program” from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was also supported in part by the Biodiversity Research Project of the Transdisciplinary Research Integration Center, Research Organization of Information and Systems.
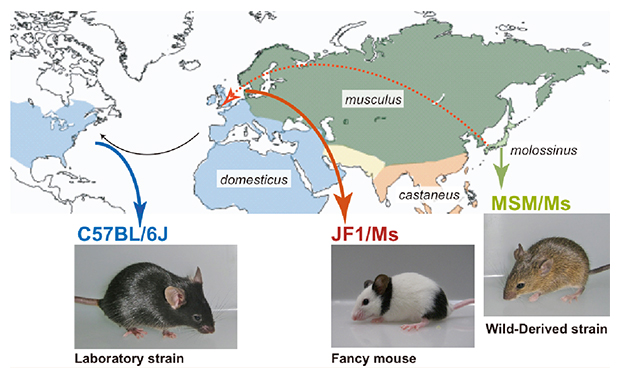
Three mouse strains mainly used in this study. C57BL/6J (B6) is a representative classical inbred strain, which was derived mostly from M. m. domesticus (west European subspecies). MSM/Ms and JF1/Ms originated from M. m. molossinus (Japanese subspecies).
Mammalian Genetics Laboratory・Shiroishi Group
Human chromosomal disorders, such as trisomy or monosomy, occur in over 1% of newborns, and are often associated with developmental failures including congenital heart disease and mental retardation. Though these disorders are likely caused by imbalance in genes involving the trisomic or monosomic region, the molecular bases are unknown in most cases.
In this study, we demonstrated that Recombination-induced mutation 4 (Rim4) is a model animal of human chromosomal disorder, Partial trisomy distal 4q (4q+). Rim4 genome has extra fragment of 6.5Mb from mouse chromosome 8, which is syntenic to the distal end of human Chr4, 4q32.3 to 4q34.1, and contains 17 genes including basic helix-loop-helix transcription (bHLH) factor, Hand2. Rebalancing the gene dosage by genetic cross with Hand2 knockout mouse rescued symptoms of the heart and limb deformities of Rim4. These results suggest that over-dosage of Hand2 causes heart and limb deformities in Rim4 and 4q+, and Rim4 provides a unique animal model to understand the molecular bases underlying the complex phenotypes of 4q+.
This work was carried out in collaboration with National Institute of Genetics (Dr. Shiroishi group) and National Hospital Organization, Niigata Hospital (Dr. Tomizawa group).
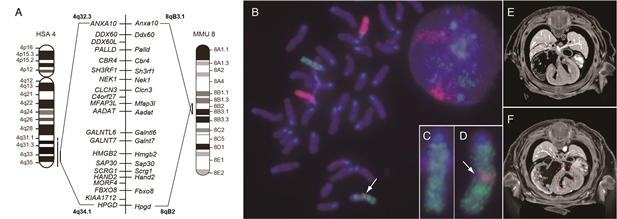
Chromosome aberration and ventricular septal defect observed in Rim4 mutant mouse.
(A) Mouse Chr8 (MMU) 8qB2–B3.1 is syntenic to human Chr4 (HSA4) 4q32.3–34.1, and genes and gene order are well conserved in the syntenic regions of the two species. (B) Dual-color whole chromosome FISH image of Chr6 (green color) and Chr8 (magenta color). Magnified images of Wild type and Rim4/+ Chr6 are shown in insets, (C) and (D), respectively. Arrow in (B) and (D) indicates Chr8-derived insertion fragment. μ-CT images of E14.5 embryos of wild type (E) and Rim4/Rim4 mouse (F) are shown. Dotted circle in (F) indicates ventricular septal defect.
Experimental Farm・Nonomura Group
The genome of cultivated rice contains 32,000 genes, and more than 20,000 are expressed in developing anther and pollen. However, there are few genes whose function is evidenced in pollen development. In this study, we succeeded to identify the rice gene with its function specifically in pollen formation, and named COLLAPSED ABNORMAL POLLEN1 (CAP1).
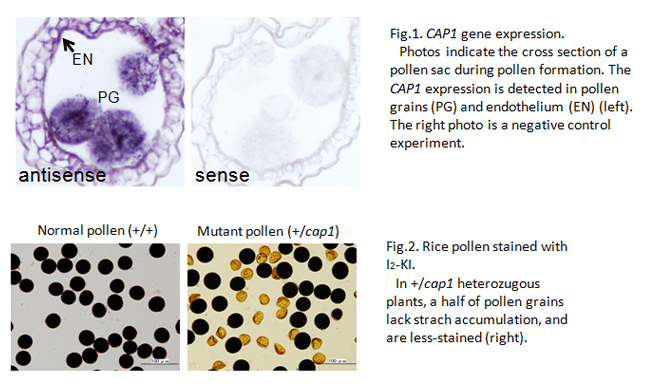
In angiosperms, the pollen has a special, tricellular structure of a pair of sperm cells involved within the vegetative cell. The CAP1 gene is strongly expressed in anthers at the bicellular pollen stage (Fig.1). The pollen grains lacking CAP1 function lose most cellular components except for outer pollen wall, exine (Fig.2), and are unable to elongate the pollen tube at all. Homozygous cap1 mutant plants exhibit no remarkable aberration in other developmental stages, indicating the specific function of CAP1 in pollen development.
The amino acid alignment of rice CAP1 protein is similar to that of the plant L-arabinokinase. The function of this enzyme is supposed in phosphorylation of free L-arabinoses, generated during the cell-wall metabolism, for reuse in de novo wall formation. It is likely in rice cap1 mutant that the arabinoses unable to be reused accumulate aberrantly or the cell-wall metabolism is disrupted in pollen grains. One of Arabidopsis L-arabinokinase-like genes shows a similar expression pattern to rice CAP1-gene expression. This result suggests that the CAP1 function is conserved broadly among angiosperm species, and plays an important role in pollen development.
This is a collaborative work with the Akita Prefectural University and the National Institute of Agribiological Sciences, Japan, and is supported by the NIG Collaborative Research Funding.
Division of Molecular and Developmental Biology・Kawakami Group
Transcription factors (TFs) are able to regulate differentiation related processes, including dedifferentiation and direct conversion, through the regulation of cell type-specific transcriptional profiles. However, the functional interactions between the TFs regulating different transcriptional profiles are not well understood. Here, we show that the TFs capable of inducing cell type-specific transcriptional profiles prevent the dedifferentiation induced by TFs for pluripotency. Of the large number of TFs expressed in a neural lineage cell line, we identified a subset of TFs that, when overexpressed, strongly interfered with the dedifferentiation triggered by the procedure to generate induced pluripotent stem cells. This interference occurred through a maintenance mechanism of the cell type-specific transcriptional profile. Strikingly, the maintenance activity of the interfering TF set was strong enough to induce the cell line-specific transcriptional profile when overexpressed in a heterologous cell type. Our results suggest that dedifferentiation suppresses a cell type-specific transcriptional profile, which is primarily maintained by a small subset of TFs capable of inducing direct conversion. We anticipate that this functional correlation might be applicable in various cell types and might facilitate the identification of TFs with induction activity in efforts to understand differentiation.
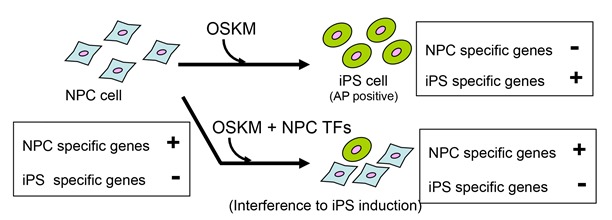
NPC (neural progenitor) cells are dedifferentiated by introduction of OSKM. This dedifferentiation was inhibited by expression n of NPC-specific transcription factors.










