



![]()
Latent environment allocation of microbial community data
Koichi Higashi, Shinya Suzuki, Shin Kurosawa, Hiroshi Mori and Ken Kurokawa
PLOS Computational Biology Published: June 6, 2018 DOI:10.1371/journal.pcbi.1006143
Pressrelease (In Japanese only)
As data for microbial community structures found in various environments has increased, studies have examined the relationship between environmental labels given to retrieved microbial samples and their community structures. However, because environments continuously change over time and space, mixed states of some environments and its effects on community formation should be considered, instead of evaluating effects of discrete environmental categories. Here we applied a hierarchical Bayesian model to paired datasets containing more than 30,000 samples of microbial community structures and sample description documents. From the training results, we extracted latent environmental topics that associate co-occurring microbes with co-occurring word sets among samples. Topics are the core elements of environmental mixtures and the visualization of topic-based samples clarifies the connections of various environments. Based on the model training results, we developed a web application, LEA (Latent Environment Allocation), which provides the way to evaluate typicality and heterogeneity of microbial communities in newly obtained samples without confining environmental categories to be compared. Because topics link words and microbes, LEA also enables to search samples semantically related to the query out of 30,000 microbiome samples.
Source: Koichi Higashi, et al., (2018), 14,e1006143, PLOS Computational Biology, DOI:10.1371/journal.pcbi.1006143
 |
 |
| College of Life Science, National Taiwan University (JENG, Shih-Tong, Dean and TING, Chau-Ti, Associate Professor) |
National Institute of Genetics (KATSURA, Isao, Director-General and KAWAKAMI, Koichi, Professor) |
Requirement of the 3′-UTR-dependent suppression of DAZL in oocytes for pre-implantation mouse development
Kurumi Fukuda, Aki Masuda, Takuma Naka, Atsushi Suzuki, Yuzuru Kato, and Yumiko Saga
Plos Genetics Published: June 8, 2018 DOI:10.1371/journal.pgen.1007436
Oogenesis is regulated by precise gene expression. One of important gene of mouse oogenesis is Dazl which has a role in translation promotion and indispensable for embryonic oocytes. Dazl is thought to be important whole life of oocyte because the expression of Dazl mRNA is detectable from embryonic to postnatal stage. In this study, we found that Dazl protein need to be suppressed in postnatal ovary whereas it has indispensable role in embryonic ovary. If this regulation does not work, female causes litter size reduction due to the defect in pre-implantation development. Thus, switching the Dazl expression from embryonic to postnatal stage by post-transcriptional regulation via Dazl’s 3’UTR is crucial for regulation production of next generation.
This study was partly supported by Grant-in-Aid for Young Scientists (B) to Y.K. (No. 25840091) and Grant-in-Aid for Scientific Research (A) to Y.S. (No. 26251025) from JSPS and by Grant-in-Aid for Scientific Research on Innovative Areas from MEXT to Y.S. (No. 25112002), A.S. (No. 16H01252), and Y.K. (No. 16H01259). K.F. is a JSPS Research Fellow (No. 16J11687).
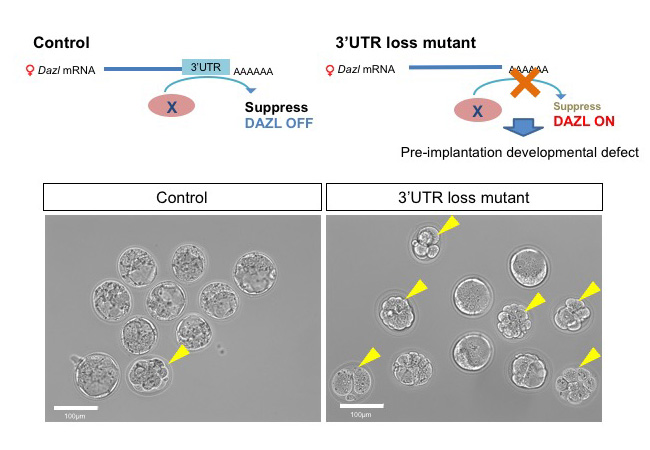
Figure: DAZL expression is suppressed in postnatal ovary in control (Upper left scheme). On the other hands, the mutant oocytes which lack 3’UTR of Dazl retain DAZL expression even in the postnatal stage and exhibit defects during preimplantation zygote stages (Upper right scheme). In control, most of all E3.5 zygotes became blastocysts (lower left panel), but in the mutant half of them stopped development (lower right panel). Yellow arrow heads indicate zygotes showing arrested development.
VITCOMIC2: visualization tool for the phylogenetic composition of microbial communities based on 16S rRNA gene amplicons and metagenomic shotgun sequencing.
Mori H, Maruyama T, Yano M, Yamada T, Kurokawa K. BMC Syst Biol
BMC Systems Biology 12 (Suppl 2), 30, 2018. DOI:10.1186/s12918-018-0545-2
Dr. Hiroshi Mori and Dr. Ken Kurokawa in the Genome Evolution laboratory from the Center for Information Biology in National Institute of Genetics, and colleagues developed a web application “VITCOMIC2” to infer taxonomic composition of a microbial community from the metagenomic sequencing data and the 16S rRNA gene amplicon sequencing data. VITCOMIC2 conducts fast and accurate taxonomic assignment of metagenomic data by using a GPU-based DNA sequence similarity search tool CLAST. Using VITCOMIC2, users can avoid the taxonomic assignment ambiguities which are derived from sequence clustering before conducting taxonomic assignment. VITCOMIC2 can be used in http://vitcomic.org .
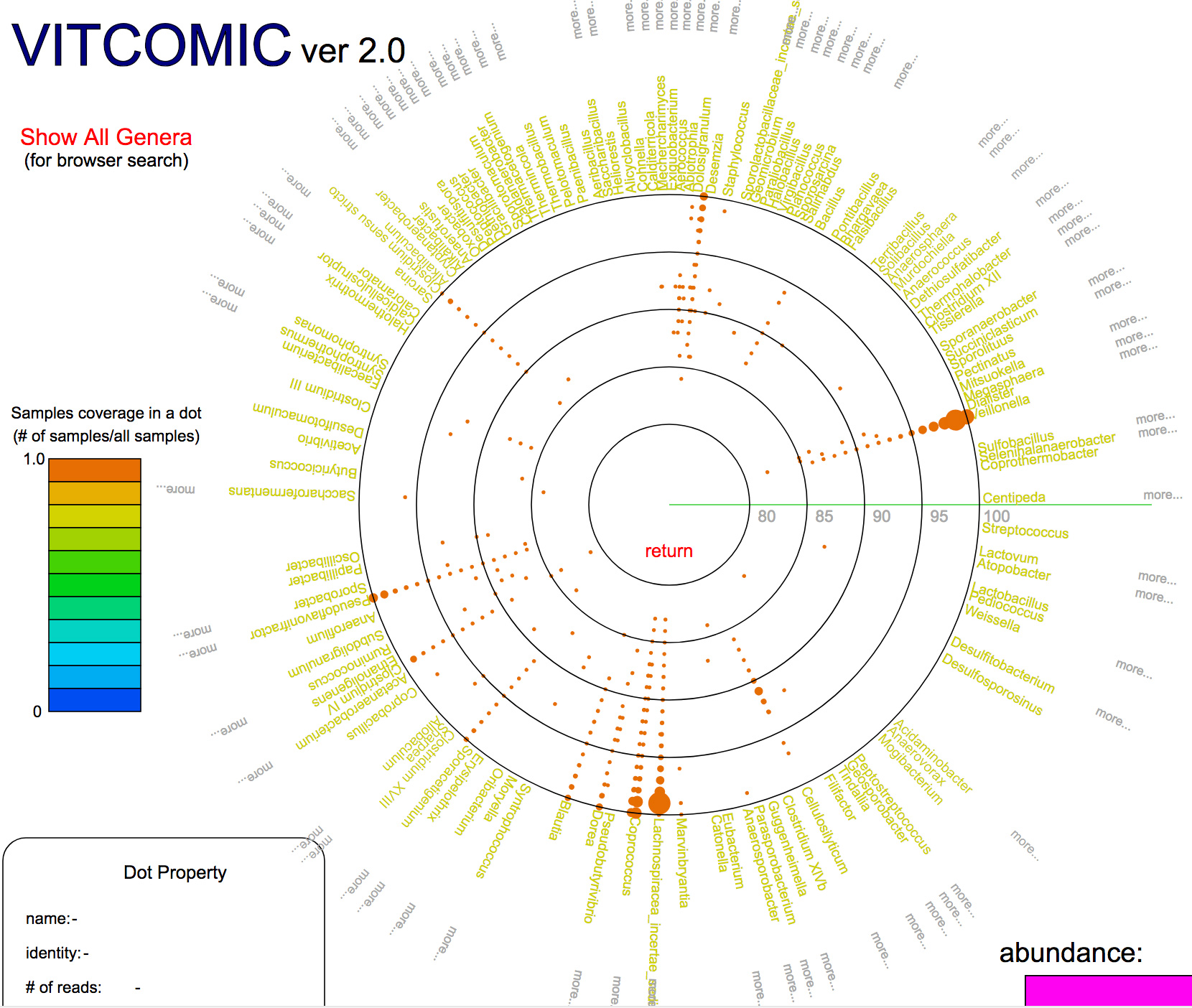
図:Example of a VITCOMIC2 result
New assistant professor joins NIG as of June 1, 2018.
Takayuki TORISAWA: Cell Architecture Laboratory • Kimura Group










